

- 904.50 KB
- 2022-04-22 11:22:23 发布


- 1、本文档共5页,可阅读全部内容。
- 2、本文档内容版权归属内容提供方,所产生的收益全部归内容提供方所有。如果您对本文有版权争议,可选择认领,认领后既往收益都归您。
- 3、本文档由用户上传,本站不保证质量和数量令人满意,可能有诸多瑕疵,付费之前,请仔细先通过免费阅读内容等途径辨别内容交易风险。如存在严重挂羊头卖狗肉之情形,可联系本站下载客服投诉处理。
- 文档侵权举报电话:19940600175。
'核磁共振项目商业计划书第八章核磁共振核磁共振(NuclearMagneticResonanceSpectroscopy)是化学领域中广泛应用的分析技术和研究工具。但由于其灵敏度在某种程度上不如气相色谱和质谱。因此,过去在农药分析上未被普遍采用。近十余年来,由于核磁共振仪器从各方面进行了改进,灵敏度有很大的提高,才逐渐推广应用。一、基本原理核磁共振谱是由具有磁矩的原子核受射频场的照射而发生跃迁所形成的吸收光谱。众所周知,原于核由质子和中子组成。其质子数与核外电子数相等,亦与原子序数相等。原子质量数为质子数和中子数之和。原子的质量数和原子序数都是偶数时,自旋量子数为零(I=0)。原子的质量数和原子序数至少有一个为奇数时,其自旋量子数才不为零(I≠0)。I≠0的原子核本身的自旋运动,将产生自旋角动量(),并使核有一个磁矩()。具有磁矩的核在静磁场H0中,就会有一定的运动和取向。除其原有的自旋运动外还会产生围绕H0的陀螺式运动即进动(见图8—1),且有自己特定的自旋量子数。、、、、等原子核,它们的质量数和原子序数均为偶数,自旋量子数I=0。它们没有磁矩,不产生核磁共振,因此,不能用于核磁共振研究。、、、、、等原子核,它们的质量数为奇数,原子序数为奇数或偶数,自旋量子数I=1/2,原子核可看作电荷均匀分布的球体,能像陀螺一样自旋,自旋的核具有循环的电荷,因而可产生磁场,形成磁矩,即μ≠0。这类核适用于核磁共振研究。自旋量子数I>1的原子核,例如(I=3/2),(I=5/2),它们的质量数为奇数,原子序数为奇数或偶数:又如:(I=1),,(I=1),它们的质量数为偶数,原子35
序数为奇数,由于自旋核具有循环的电荷,能产生磁场,形成磁矩,即μ≠0,由此都可适用于核磁共振研究。(见表8—1)。以上所列原子核,多可供核磁共振研究,其中以1H最容易测出,因此,目前农药分析中最常用的是1H—NMR的测定也是本章的重点。其次,用的较多的是13C、31P和19F。虽然13C的天然丰度很小,只有1.069%(1H为99.9844%,19F为100%,31P为100%)。且其信号灵敏度只有质子的1/63,较难测定。但近年来,由于仪器和操作技术的改进,测定13CNMR谱在结构测定中已占十分重要的地位。在磁场中,各种核所产生的磁矩有一定的取向,由磁量子数(m)决定,而磁量子数m由核的自旋量子数决定,即:m=I,(I-1),(I-2),…,-I由此,共有(2I+1)个m值。1H,13C等核,其I=1/2,则只可能有两种取向,即m=+1/2,表示核磁矩顺着H0方向(↑)m=-1/2,表示核磁矩逆着H0方向(↓)。质子磁矩的两种取向相当于两个能态。磁矩方向与磁场相同(顺H0方向)的,质子能态低,不相同(逆H0方向)的,质子能态高。若以射频场照射磁场中的质子,当射频场的能态与两个能态的能量差相等时,处于低能态的质子就可吸收射频场的能量跃迁到高能态。这就是核磁共振,上述两个能态间的能量差可以下式表示:式中:h——普朗克(Plank)常数h=6.626176±0.000036×10-34J·sv——共振频率。共振频率和外磁场强度之间又有如下的关系:式中:γ——磁旋比,即核的磁矩与角动量的比值,是核固有的性质;H0——外磁场强度。对于相同的原子核,γ为常数,不同的原子核,则γ不同。由此,改变外磁场强度H0或改变辐射能频率v都可保持上式的关系。目前,一般核磁共振仪多采用固定辐射频率而改变磁场强度H0的方法,更便于获得能量吸收曲线,即核磁共振谱图。见图8—2。35
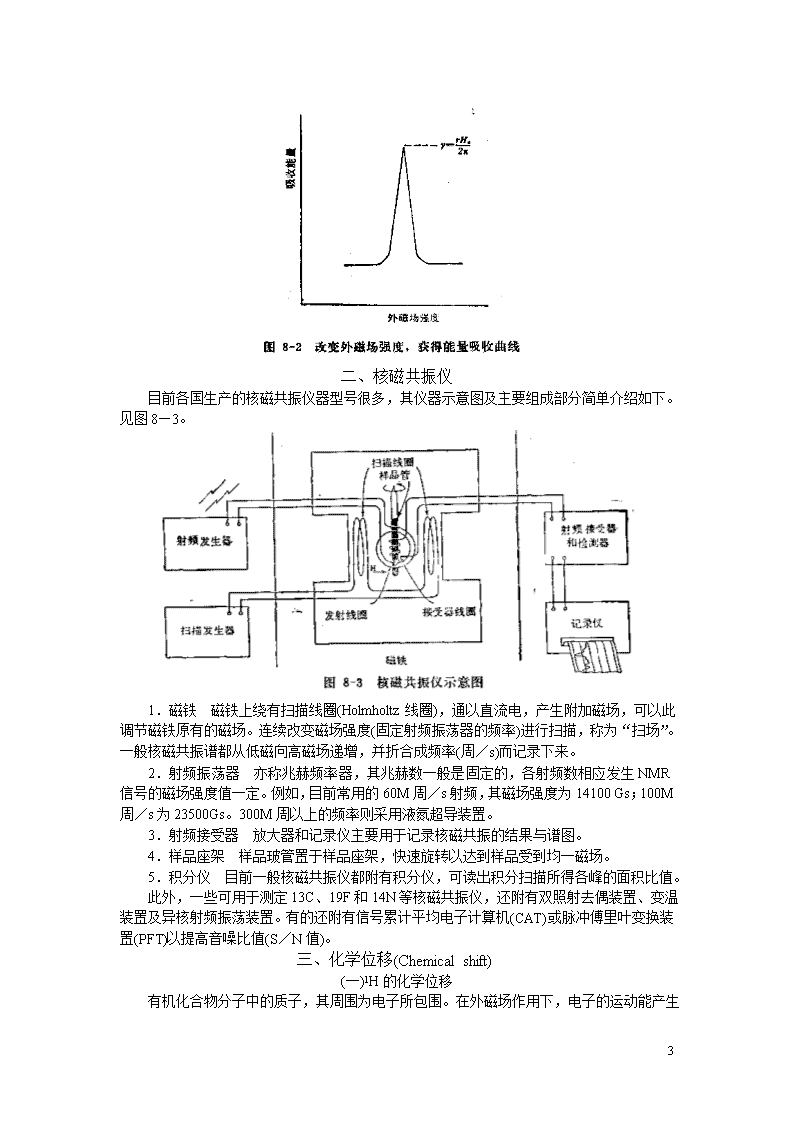
二、核磁共振仪目前各国生产的核磁共振仪器型号很多,其仪器示意图及主要组成部分简单介绍如下。见图8—3。1.磁铁磁铁上绕有扫描线圈(Holmholtz线圈),通以直流电,产生附加磁场,可以此调节磁铁原有的磁场。连续改变磁场强度(固定射频振荡器的频率)进行扫描,称为“扫场”。一般核磁共振谱都从低磁向高磁场递增,并折合成频率(周/s)而记录下来。2.射频振荡器亦称兆赫频率器,其兆赫数一般是固定的,各射频数相应发生NMR信号的磁场强度值一定。例如,目前常用的60M周/s射频,其磁场强度为14100Gs;100M周/s为23500Gs。300M周以上的频率则采用液氮超导装置。3.射频接受器放大器和记录仪主要用于记录核磁共振的结果与谱图。4.样品座架样品玻管置于样品座架,快速旋转以达到样品受到均一磁场。5.积分仪目前一般核磁共振仪都附有积分仪,可读出积分扫描所得各峰的面积比值。此外,一些可用于测定13C、19F和14N等核磁共振仪,还附有双照射去偶装置、变温装置及异核射频振荡装置。有的还附有信号累计平均电子计算机(CAT)或脉冲傅里叶变换装置(PFT)以提高音噪比值(S/N值)。三、化学位移(Chemicalshift)(一)1H的化学位移有机化合物分子中的质子,其周围为电子所包围。在外磁场作用下,电子的运动能产生35
感应磁场,由此,质子所感受到的磁场强度并不完全等于外磁场强度。质子周围的电子使质子实际感受到的磁场强度往往比外加磁场强度要弱,这就是说,电子对外磁场有屏蔽作用。其屏蔽效应的大小一般决定于质子周围电子云密度的高低。电子云密度愈高,屏蔽效应愈大,该质子的信号需在更高的磁场强度下才能获得。反之,电子和外磁场平行排列,增加了外磁场强度,质子就受到去屏蔽作用。该质子的信号只需在较低的磁场强度下就能获得。上述两种效应分别称为抗磁屏蔽效应和顺磁屏蔽效应。在1HNMR测定中,化合物中的各种氢核,处于不同的化学环境之中,它们都受到核外电子的屏蔽,这些核外电子的密度又受其邻近原子的电负性、原子间键的性质以及杂化等多种影响,屏蔽程度亦各不相同,由此,不同的1H,其共振频率都各有差异。它们实受到的磁场往往小于H0。各种不同化学环境的1H共振频率相差不大,如在100MHz的仪器上1H的共振频率的差别约为1500Hz。由于无法以裸核作标准测出其绝对值,所以选用一种标准物质,以标准物质的共振峰与测定化合物中的各种1H的相对距离Hz数,作为这些1H的化学位移。由此,化学位移的定义可以下式表示:δ为化学位移,v为频率,单位为周/s。由于磁场强度与共振频率成正比关系,上式亦可表示为:由于标准的共振频率数目较大,且与辐射频率相差不多,为方便起见,即以v辐射频率代替v标准,单位为周/s。因数值太小,为便于计算,各乘以106并设单位为ppm(partpermillion)。最常用的标准物质为四甲基硅烷(CH3)4Si,简称TMS。由于Si的电负性低,四甲基上氢的屏蔽效应大.它的共振信号在高场位,且为强而锐的吸收峰。以TMS的共振峰为零(δTMS=0)。共振峰的共振频率与TMS相差60周/s时,δ为1;相差120周/s时,δ为2;依此类推。核磁共振谱图中,常以化学位移δ为横坐标表示。化学位移也有用τ表示的。τ的定义为10—δ=τ。例如,δ=2,则τ=8。用δ表示时,0是高场;用τ表示时,10是高场。有些化合物的质子信号,发生在δ=0以下,δ是负数,表示屏蔽效应很强;反之,δ=10以上,表示去屏蔽效应很强。以δ表示化学位移,不因仪器不同而有所差异,使用方便,现为国际上采用。下面列举一些基团中质子的化学位移(δ值)。见表8—2。8—3,8—4。35
35
(二)13C的化学位侈13C的化学位移范围很宽。一般有机化合物中各种碳原于的δc值范围在0一250ppm。一个不对称的分子,几乎每个碳原于都有各自的δc峰。从下列各表中,可以看到不同碳原子所处环境不同,其化学位移亦各不相同。δc一般也以TMS为内标物。δTMS=0ppm。下面列举—些化合物类型的δc:(1)(取代)三元环、C—Si、长链—烷烃:0—10ppm或15ppm;(2)烷烃、CH2或CH3上连有O、N、S、Br、C1:30—50ppm;(3)环氧化合物、或CH一连有Br、Cl:50—70ppm;(4)炔烃、CH2上连有F:70一90ppm;(5)-CCl3、-CHF、O-C-O:90—105ppm;(6)(取代)芳烃、杂芳烃、烯烃、:105—150ppm;(7)羰基化合物(羟酸、盐、酯、酰胺等),肟:150一180ppm;(8)不饱和醛或酮、不饱和:180-200ppm;(9)饱和酮、饱和、正碳离子:>200ppm。此外,表8-5和表8-6还分别列出一些烯烃和杂芳环化合物的13C化学位移。35
(三)31P的化学位移31P—NMR具有较强的信号,因为31P的丰度较大。31P-NMR一般以85%H3PO4为内标物。δp—0,由于31P谱线较宽;且δ随温度而变化,因此,一般报道的数据有5pm的出入。下面列举一些31PNMR的数据。见表8—7。四、自旋-自旋偶合和自旋—自旋裂分有机化合物的核磁共振谱图中,有些质子的吸收峰不是单峰,而是一组双峰、三峰或多重峰。这是由于磁核之间的相互作用所引起的能态裂分,即一个质子受到邻近质子的自旋状态以及所产生的感应磁场的影响导致的裂分现象。这种相互作用称自旋—自旋偶合(Spin—spincoupling)。其作用的大小可用J来表示。J为自旋—自旋偶合常数,简称自旋偶合常数(Couplingconstant),单位为Hz。自旋—自旋偶合所产生的裂分现象称为自旋—自旋裂分(Spin-spinsplitting),简称自旋裂分。如果外磁场为H0,相邻的一个质子可增强外界的场力使总磁场强度为H0+H’=H1;如相邻的一个质子减弱外界的场力,则总磁场为H0-H’=H235
。由此,一个质子发出的信号就裂分为两个。这种自旋—自旋裂分系由于该质于和邻近质子自旋—自旋偶合而产生的。若一个质子受邻近一个质子的影响,则裂分为两个信号,即产生二重峰,其信号强度为1∶1。若一个质子受邻近两个质子的影响,则裂分为三个信号,即产生三重峰,其信号强度为1∶2∶1。若一个质子受邻近三个质子的影响,则裂分为四个信号,即产生四重峰,其信号强度为1∶3∶3∶1。由此,可见偶合裂分的峰数,有一定的规律,一组等同的质子,可使一个邻近的质子发出的信号裂分为n+1个峰。下面举两个例子说明自旋—自旋偶合和自旋—自旋裂分现象。[例1]3-戊酮从图8-4可以看出,A为三重峰,系分子中CH335
质子受相邻CH2的影响而裂分为三重峰,而B为四重峰,系分子中CH2质子受相邻CH3的影响而裂分为四重峰的。[例2]乙醇(CH3一CH2一OH)乙醇分子中有三类不同的质子,因此,其核磁共振谱图(图8—5)中出现三组峰。A组峰系乙醇分子中CH3上的质子与相邻CH2质子偶合裂分形成的三重峰,C组峰系OH上的质子与相邻CH2上的质子偶合裂分为三重峰,而乙醇分子中CH2上的质子、由于和相邻CH3上的质子偶合,又与相邻OH上的质子偶合,因此,第一次偶合形成的四重峰,再进行一次偶合,裂分为双四重峰,从而形成一个分辨很不甚清楚的八重峰(B组峰)。必须注意的是一个质子只能和化学位移不同的相邻质子发生偶合和裂分。凡两个质子被三个键分隔的,例如,都可发生偶合裂分,但相隔三个键以上的质子,一般都观测不到偶合和裂分。同一个碳上的质子,除非环境不同,两个质子不等同,一般不发生偶合裂分。此外,还必须注意偶合裂分后峰与峰之间的距离。这种裂分距离即偶合常数,是测定两个质子偶合效力的标准。偶合常数与外磁场强度无关,仅取决于分子的结构和几何形象,且与核上电荷密度及两个相偶合的核所处的相对位置有关。偶合常数大,表示偶合效果好。因此,偶合常数也是物质结构、构型、构象的重要信息之一,尤其在较复杂的分子中,偶合常数的测定显得更为重要。35
下面列举一些质子—质子的偶合常数。见表8—8和表8—9。35
五、决定质子数目的方法在核磁共振谱图上信号的面积与各质子的数目成正比例关系,由此,将各信号的面积进行比较,即可决定各35
质子的相对数目。信号面积一般用阶梯式积分曲线表示,积分线由低磁场画向高磁场。自积分线的出发点至终点的全高度与所有质子的数目成正比,每一阶梯的高度与相应的质子成正比。其高度可按记录纸上的小方格为单位表示。例如,某分子有7个质子,其核磁共振谱图(图8—6)上积分线总高度为14小格,则每一格相当于0.5个质子(即7个H/14格=0.5个H/格)。A处含4格,相当于2个质子;B处含8格,相当于4个质子;C处含2格,相当于1个质子。由此,质子数分别为2∶4∶1。六、在农药分析中的应用(一)在农药分析中的应用范围在农药分析研究中,核磁共振可以从以下多个方面提供特殊的信息;(1)农药分子结构的鉴定和确定;(2)农药纯度的分析和控制;(3)农药化合物分子的几何异构体、(4)农药分子内电荷的分布;(5)农药分子间的相互作用,络合物的形成以及键合的倾向;(6)农药分子间的运动、交换和化学平衡;(7)农药分子的降解和生物代谢。(二)农药样品的准备农药样品的核磁共振测定,必须在具盖的核磁共振样品玻管中进行。核磁共振样品管要求玻璃透明度好,且同心度和正直度高,有利于在磁场中均匀旋转。样品管具有一定规格和直径,一般可分为1.7,2.5,5,10,15mm等多种规格,其中最常用的为5mm和10mm的样品管。样品管必须事先洗净、干燥、备用。液体样品可直接装入核磁样品管,加入内标物进行测定。一般样品用量为0.4ml或含样品5—20mg即可。固体或粘稠液体样品必须加溶剂溶解后方可进行测定。所需溶剂要求对样品的溶解度好,最好本身不含氢,以避免峰的干扰。常用的溶剂为四氯化碳、二硫化碳、三氯一氟甲烷(CCl3F)和三氟乙酸(CF3COOH)等。必要时需用重水(D2O)或氘代有机溶剂,例如,氘代氯仿(CDCl3),氘代甲醇(CD3OD),氘代丙酮(CD3COCD3),氘代二甲亚砜(),氘代三氟乙酸(CF3COOD),氘代吡啶、氘代苯、氘代二甲基甲酰胺、氘代乙腈(CD3CN)等。由于氘代有机溶剂价值昂贵,需节约使用。(三)内标物1H和13C—NMR,一般采用四甲基硅烷(CH3)4Si(简称TMS),为内标物。它是沸点27℃的化学上很惰性的无色液体,易于挥发,可配成10%-20%的CCl4溶液,贮于磨口瓶中备用。35
高温时,采用六甲基二硅烷(HMDS)或六甲基二硅醚(HMDSO)为内标物。它们是沸点高于100℃的无色液体。有时也采用DSS[(CH3)3SiCH2CH2CH2SO3Na]作为内标物,由于它是水溶性的,以D2O作溶剂时,用DSS十分适宜。但必须注意,它本身有许多谱线,用量切勿过多,以免在0.5—3.0ppm范围内出现干扰峰,有人建议用(CH3)3SiCD2CD2CD2Na代替DSS,可避免上述范围内三个亚甲基峰的干扰。(四)农药样品的核磁共振测定下面列举一些农药样品的核磁共振测定。1.毒死蜱的结构测定毒死蜱(Chlorpyrifos)是一种广谱杀虫剂,可用于防治蚊、蝇等卫生害虫、各种土壤害虫和许多叶类作物害虫。纯品为熔点42.5—43℃的白色结晶,化学名称为O,O—二乙基—O—3,5,6—三氯—2—吡啶基硫逐磷酸酯。化学结构式为:毒死蜱溶于氘代丙酮,以TMS为内标物进行1H—NMR测定。其核磁共振谱见图8-7。由上谱可见δH1.4l是CH3基的核磁共振峰、它与CH2基偶合而裂分为三重峰。三重峰中每一个峰又受31P核的远程偶合的影响而进一步裂分。δH4.40是CH2基的核磁共振峰,它与CH3基偶合而裂分为四重峰,并受31P的影响而进一步裂分。31P(I=1/2与乙基质子通过氧原子连接,其偶合常数比31P与乙基质子直接相连的要小得多。它们分别为JP-CH3=0.9Hz和JP-CH2=2.0Hz。δH8.3l为芳烃质子的核磁共振峰。由于它与31P核偶合而裂分为二重峰。0ppm处为TMS内标物的核磁共振。一个宽峰为OH峰,可能是样品中有残留的水。此外,还有末完全被氘代的丙酮五重峰。2.鱼藤酮的鉴定和纯度测定鱼藤酮(Rotenone)是由鱼藤属植物根部提取获得的一类天然杀虫剂。可用核磁共振方法对鱼藤酮进行鉴定和纯度确定。从核磁共振谱图(图8—8)中可以看出样品A和B的峰形相似,而样品C的谱图中(芳烃35
区)还出现额外的峰,说明这个样品含有杂质。此外,样品C的谱图中,一些峰的位置有所偏移。例如,样品A和B的甲氧基核磁共振峰,分别为δH3.07和δH3.764,而C则为δH3.392和δH3.255。甲基和芳烃质子峰也有偏移,它只能是A和B的异构体或分解产物。3.马拉硫磷的降解和反应马拉硫磷(Malathion,马拉松)是一类非内吸广谱性杀虫剂,有良好的触杀和一定的熏蒸作用,适用于防治烟草、茶、桑树等的害虫及仓库害虫。35
在生物体内和有水的环境中,它被降解为马拉硫磷的单酸(α-和β-单酸)。在13CNMR谱(图8—9)上,可以见到13C的化学位移和13C—31P的偶合常数。此外,1H—NMR可用来测定马拉硫磷在33%甲醇水溶液和羟胺水溶液中的消除、羟胺化和乙酯化反应的相对速度,4.二氯代萘代谢产物的确定根据1H—NMR测得二氯代萘的代谢产物为二氯代萘酚,但尚无法测知该代谢产物是2,6—二氯—l—羟基萘(A)还是l,6—二氯—2—羟基萘(B)。经13CNMR的测定,按照图8—10所测得的参数,结构A的平均偏差为1.95ppm,而结构B的平均偏差为0.47ppm。由此,可知二氯代萘的代谢产物为l,6—二氯—2—羟基萘(B)。第九章滴定分析法滴定分析是将已知浓度的试剂溶液(或称标准溶液)从滴定管滴加到待测物质的溶液中,直至标准溶液与被测组分按化学计量反应完全,根据标准溶液的浓度,计算被测物质的含量,反应完全的这一点称为等当点。指示剂发生颜色变化的转变点为滴定终点。根据滴定终点可以确定等当点,但滴定终点与等当点不完全符合,两者之差称为终点误差。滴定分析法较准确,操作简便快速,仪器设备简单,在广泛应用光谱法和色谱法以前,农药分析中主要使用滴定法和重量法,重量法目前用得很少,滴定法仍然使用。35
根据所利用的化学反应不同,在农药分析中常用的滴定分析法有:①酸碱滴定法;②氧化还原滴定法;③沉淀滴定法;④亚硝酸钠滴定法;⑤非水滴定法。在不使用指示剂的其它指示终点的滴定分析方法,如电位滴定法、永停滴定法在农药分析中亦有应用。滴定分析法是利用农药有效成分中某元素或原子团与标准溶液起化学反应作为计算有效成分含量的依据,适用于测定纯度较高的农药。如果农药纯度较低,其中杂质亦会产生相同反应时,误将杂质亦算作农药,使测定结果不准确,这样的例子是很多的,如杀螟硫磷乳油的测定,早期各工厂都用直接重氮化法,后将此法与气相色谱法进行对比,结果列于表9—l。说明直接重氮化法测定杀螟硫磷含量,一般比气相色谱法的结果高出6—8个百分点,因此确定气相色谱法为标准方法。长期使用直接铜盐比色法测定马拉硫磷,结果偏高,改用薄层铜盐法才符合要求;又如采用碱解法测定50%辛硫磷乳油,原油中有能与碱反应的杂质,使测定结果偏高。样品经薄层分离后,用极谱法测定或直接使用高效液相色谱法测定,其含量只有35%左右,因此原药纯度低的农药样本不能直接使用滴定法,目前许多农药已经采用具有分离杂质手段的测定方法,如气相色谱法和高效液相色谱法,但有的农药在气谱条件下易分解,液谱仪价格昂贵,在一般工厂质量控制中,利用薄层色谱首先将农药与杂质分离,将薄层板上的农药洗脱下来进行化学测定,可以得到满意的结果。一般相对偏差在1.0%以下。此外、薄层—滴定法测定时不需要农药标准品,根据化学反应消耗的试剂可以直接算出有效成分的含量。我国农药国家标准中。使用此法的有甲胺磷薄层碘量法(GB372—83)乐果的薄层溴化法(HG2—l417—81),矮壮素的沉淀滴定法(HG3—818—75)(1983确认),氨基甲酸酯类农药异丙威(GB9560—88)和速灭威(GB—6592—88)含量的测定均使用薄层定胺法和气相色谱法,但以薄层定胺法为仲裁法。一、容量分析法(一)酸碱滴定法是以酸碱反应为基础的滴定分析法、选用适当的有机弱酸或弱碱作指示剂,它们在不同酸度下呈现不同的颜色,常见的酸碱指示剂如表9—2。1.农药酸度的测定很多农药特别是磷酸酯类、拟除虫菊酯类、氨基甲酸酯类农药等在碱性条件下会分解,因此酸度是农药质量的一个重要指标。在我国,农药制剂的酸度通常用pH表示;原药以酸含量表示,即以样品中所含硫酸或盐酸的质量百分数来表示,测定方法可参阅第十一章“酸度测定。”2.农药含量测定敌敌畏经薄层色谱分离杂质后,在0-1℃用1mol/L氢氧化钠标准溶液反应20min,可定量地水解成二甲氧基磷酸钠和二氯乙醛,用标准酸溶液滴定反应后剩余的碱,根据消耗的碱量计算其含量。35
如果水解条件变化,二氯乙醛在碱性溶液中可继续分解而消耗碱液,因此测定时必须严格控制时间和温度。Cl2CHCHO+2NaOH→HCOONa+HCHO+2NaCl+H2O氨基甲酸酯农药如异丙威、速灭威等,经薄层分离后,可通过碱解反应,定量的放出挥发性甲胺,用硼酸吸收。再用盐酸标准溶液滴定,从而计算出有效成分含量,这类农药的国家标准荡薄层定胺法是中和法。CH3NH2+H3BO3→CH3NH3H2BO3CH3NH3H2BO3+HCl→H3BO3+CH3NH3Cl滴定时不能用酚酞,只能使用甲基红或溴甲酚绿作指示剂。苯基脲类除草剂如敌草隆,在碱性溶液中水解出二甲胺,亦可使用蒸馏定胺法,水解产物二甲胺蒸入硼酸溶液中,用标准盐酸滴定二甲胺,计算敌草隆含量。终点指示剂可使用电位法,甘汞电极一玻璃电极对,也可选用指示剂。(二)氧化还原滴定法是以氧化还原反应为基础的滴定分析法。使用本方法有三点要求:①反应能定量地完成;②反应速度快;③有较简便可靠的方法确定等当点。用于分析农药的方法有;碘量法、溴酸钾法、高锰酸钾法等,这些方法的特点是反应时间短,操作简单,终点明显,空白值小。但如溴化法与薄层色谱法联用时,展开剂甲醇、乙醇、丙酮、石油醚等都可能消耗溴而有干扰、必须尽量除去展开剂或选用干扰小的展开剂。在进行氧化还原反应时.农药本身的官能团有时也会干扰,测定时必须严格遵守温度、反应时间和氧化剂的量。在处理氧化还原滴定结果时,以其摩尔质量(分子量(式量)/n)作为氧化还原反应的基本单元,n为一式量被测物质在滴定过程中得到或失去的电子数。1.碘量法是利用I2的氧化性和I—的还原性进行滴定的方法。35
其摩尔质量为l/2I2=126.92,含有不饱和烃或水解后生成一SH键的组分都可试用碘量法,可分为直接碘量法和间接碘量法。直接碘量法是利用碘标准溶液直接滴定还原物质的方法,如早期(1983年)甲胺磷的国标采用薄层—碘量法,先用薄层法将甲胺磷与杂质分离,经甲酵氢氧化钠碱解后,用标准碘溶液滴定至浅黄色,碘在此处为置换反应。久效磷和磷胺亦可使用薄层—碘量法,薄层展开后,加入过量碘液反应后,用硫代硫酸钠回滴多余的碘,已知反应过程中消耗2个I2,其反应式推测可能为以下两种:有机氮农药杀螟丹可采用碘量法测定,它在碱性溶液中分解为二氢沙蚕毒,在酸性介质中可被碘氧化为沙蚕毒。加入淀粉指示剂用碘滴定至微蓝色。二硫代氨基甲酸酯类农药如代森锌、代森铵等都可用此法,这类农药遇酸能分解释放二35
硫化碳,用甲醇氢氧化钾吸收后生成黄原酸钾,然后用标准碘液滴定至淀粉指示剂变蓝,为了避免受氧气的影响而产生误差,必须立即滴定。此外,测定样品水分的卡尔费休法,也是一种碘量法。间接碘量法是利用I—离子的还原作用与氧化性物质反应生成游离的碘。再用硫代硫酸钠标准溶液滴定后测出农药的含量。砷酸药剂就是利用此法测定。在样品中加入盐酸和碘化钾溶液,碘化钾将砷还原,生成的碘用硫代硫酸钠回滴。AS2O5+4H++4I-→AS2O3+2I2+2H2O进行间接碘量法时,由于碘易挥发,在酸性溶液中I—易被空气氧化,反应应在碘量瓶中进行,同时加入过量的KI与I2生成13-以减少挥发,在室温下进行反应,温度不能太高,不要过度摇动,以避免与空气接触。下面将讨论溴酸钾法和高锰酸钾法,它们亦可算作间接碘量法。2.溴酸钾法是农药分析中应用最广的氧化还原滴定法。用过量的溴酸钾和溴化钾作标准溶液,在酸性条件下析出溴,溴与被测农药反应,剩余的溴再与碘化钾反应析出碘,用硫代硫酸钠滴定,其反应为BrO3-+5Br-+6H+→3Br2+3H2OBr2(剩余量)+2I-→2Br-+I2I2+2S2O32-→2I-+S4O62-多数硫代磷酸酯如乐果、氧乐果、久效磷、磷胺、对硫磷、杀螟硫磷等,取代硫脲、沙蚕毒素类农药在一定条件下能定量地被溴氧化,一些水解后生成酚或苯胺的农药也能定量地被溴取代。有些农药分子中的基团与溴不定量反应。如稻瘟净,则不能使用此法。溴化反应机理比较复杂,各种农药亦不相同,当反应时温度、时间及氧化剂数量改变,反应的结果亦不相同,因比必须严格遵守方法的测定条件,农药的溴化反应有以下几类:①硫逐磷酸酯()被氧化为硫酸,此反应比较迅速,在室温下,氧化剂过量20%以上,超过5min,反应即可完成。②硫赶磷酸酯上的硫()较难氧化,但不同品种各不相同,氧化乐果较易断开,溴化温度30℃较好,氧化剂必须过量;伏杀硫磷不易断开,不能用溴酸钾法,改用高锰酸钾法。③有的农药可能生成亚砜()或砜()。④组分中双键和苯环也能与溴反应,如速灭威、对硫磷等;⑤35
其它含硫农药如福美锌、福美双、杀虫双、杀螟丹等,现将几类典型农药的反应式及氧化数叙述如下。(1)乐果和氧乐果:乐果的氧化数为14,氧乐果的氧化数为6,溴化法曾被作为乐果原油和乳油标准中的分析方法。(2)对硫磷和甲基对硫磷:①对硫酸先碱解、后溴化,其氧化数为8+4=12甲基对硫磷水解后除进行(Ⅱ)式外,(Ⅲ)式中在苯环上只取代一个溴:因此氧化数为10。如果温度较高或放置时间较长,苯坏将进—步被溴代,可使结果偏高。②不碱解,在酸性介质中溴化,其氧化数为8:杀螟硫磷在苯环上比对硫磷多一个甲基,一般在相同条件测定;有时苯环上的甲基被氧化,使测定值变高,因此必须控制条件。(3)喹噁硫磷的氧化数为l0:35
(4)甲拌磷分子中的硫醚结构,在一定条件下能定量地氧化成亚砜,氧化条件加强,可一步氧化成砜,必须严格掌握氧化成砜的反应条件,其氧化数为20:(5)乙酰甲胺磷氧化数为6:(6)氨基甲酸酯农药速灭威水解后的间甲酚,可被溴定量取代,氧化数为6:(7)杀虫双氧化数为12:(8)福美双氧化数为30:35
(9)甲基硫茵灵氧化数为16:溴酸钾法也是一种间接碘量法,由于KBrO3—KBr溶液比较稳定、实验误差主要来源于滴定碘的过程中,在碘量法中已讨论过,如果是薄层溴化法,板上的溶剂一定要完全挥发掉,因为醇、酮等溶剂与溴发生反应,会影响测定结果,农资部门王全忠等用薄层溴化法对十几种农药进行分析测定,一般偏差在1%以下。3.高锰酸钾法曾用于多种无机农药的测定,现在还用于磷化锌、磷化铝的测定,把磷化锌与稀酸作用放出磷化氢气体,通入定量的高锰酸钾标准溶液中,使其氧化成磷酸,加入过量的草酸标准溶液以还原剩余的高锰酸钾,最后再用高锰酸钾标准溶液回滴多余的草酸,高锰酸钾自身为指示剂,根据高锰酸钾消耗量可计算出磷化锌含量,反应式如下:Zn3P2+3H2SO4→3ZnSO4+2PH3↑5PH3+8KMnO4+12H2SO4→4K2SO4+8MnSO4+5H3PO4+12H2O2KMnO4+5H2C2O4+3H2SO4→2MnSO4+K2SO4+8H2O+10CO2反应中通常使用硫酸而不用硝酸,因为硝酸是氧化性酸,可能与农药反应,亦不用盐酸,因盐酸中的Cl—有还原性,能与高锰酸钾反应。有的有机磷农药中的硫较难溴化,而且分子中有能和溴反应的部位,则不能使用溴酸钾法,如稻瘟净中的硫很难溴化,溴化时间长,则苯环上要起副反应,且不定量,改用高锰酸钾法,则硫易被氧化,其苯环上亦不起副反应。反应后在过量高锰酸钾中加入碘化钾,生成的碘再用硫代硫酸钠回滴至淀粉指示剂的蓝色消失。杀虫双经薄层分离后,亦可用高锰酸钾氧化,分子中两个硫原子被氧化成硫酸,根据高锰酸钾消耗量,求出杀虫双含量。35
4.氯胺T法氯胺T是对甲苯磺酰氯胺钠,在酸性介质中能氧化硫代磷酸酯农药的键和其它农药的≡C-S-键,将硫氧化成硫酸,多余的氯胺T则与碘化钾作用析出碘,可用碘量法定量。如测定治螟磷(苏化203)可用该法。(三)沉淀滴定法沉淀滴定法是以沉淀反应生成难溶物为基础的滴定分析法,能满足以下要求的沉淀反应,才能用于此种滴定分析:①反应速度快、生成沉淀的溶解度小;②应定量进行;③有准确确定等当点的方法。由于以上条件限制,能用于沉淀滴定的反应较少,微溶性银盐的沉淀反应是最常用的,以这类反应为基础的沉淀滴定法称为银量法,合有氯、溴、碘的农药可以采用此法,但不同分子结构的农药脱卤素的方法亦不同,脂肪链上的卤素,如敌百虫、敌敌畏、磷胺、滴滴涕和矮壮素等在水解脱氯时,使用氢氧化钾或氢氧化钠水溶液,敌敌畏的水解脱氯反应如下:如矮壮素[CH2Cl-CH-N+(CH3)3]Cl-国家标准中的分析方法是在碱水解后滴定总氯量后与水解前滴定游离氯之差来计算有效成分含量的,至今国际农药分析协作委员会仍应用此法。三氯杀螨醇、甲草胺等则使用氢氧化钠(或钾)的乙醇溶液水解脱氯:35
稳定直链上的卤素,如溴氰菊酯、氯氰菊酯、氯菊酯等使用氢氧化钾-乙二醇溶液脱氯:苯或杂环上的卤素、如氰戊菊酯、杀虫脒、百菌清、除草醚、敌草隆等农药,则需用金属钠在无水乙醇中回流脱下,以氰戊菊酯为例,测定样本时经薄层分离后,用二甲苯将组分洗入50m1圆底烧瓶中,加0.5g金属钠后回流,沸腾后由上口分3—4次加入10m1无水乙醇,回流约30min,用水、丙酮洗人烧杯中,在这类农药测定前需加入甲醛溶液,以去除一CN基的干扰。现以敌百虫为例说明沉淀滴定法:脱下的卤素用硝酸银标准溶液滴定,可以采用电位法,通过电位法滴定曲线指示终点,亦可用佛尔哈德法(Volhard),是用铁铵矾[NH4Fe(SO4)2]作指示剂的银量法。在农药分析中常用返滴定法测定卤素离子,以测定C1—为例,加入已知过量的硝酸银,使C1—全部形成AgCl,以铁铵矾为指示剂,用硫氰酸铵(NH4SCN)标准溶液返滴过量的银,滴入的NH4SCN首先与Ag+发生反应,生成AgSCN。在Ag+与SCN—反应完全后,过量一滴NH4SCN溶液便与Fe3+反应,生成红色的FeSCN2+络合物,指示终点已到,这时若再滴入NH4SCN时,由于AgCl的溶解度比AgSCN大、过量的SCN—将与AgCl反应,使AgCl沉淀转化为溶解度更小的AgSCN,溶液中出现红色后,如仍不断摇动溶液,红色又逐渐消失,会误认为终点未到而造成误差。Ag++Cl-→AgCl↓(白色,溶解度常数=1.8×10-10)Ag++SCN-→AgSCN↓(白色,溶解度常数=1.0×10-12)AgCl↓+SCN-→AgSCN↓+Cl-为了避免误差,在试液中加入过是的AgNO3后,加入l一2ml邻苯二甲酸二丁酯或硝基苯,用力摇动使AgCl沉淀的表面上覆盖一层有机物,避免沉淀与外部溶液接触,阻止NH4SCN与AgCl发生转化反应。用返滴定法测溴化物或碘化物时,由于AgBr及AgI的溶解度均比AgSCN小,不会发生上述转化反应。(四)亚硝酸钠滴定法亚硝酸钠滴定法又称重氮化滴定法,在农药分析中常用于测定含氨基和硝基的芳香化合物。其原理是利用芳香族伯胺类农药,在盐酸存在时能与亚硝酸作用,生成芳香族伯胺的重氮盐,其反应式为:AR-NH2+NOCl→AR-NH-NO+HClAR-N=N-OH+HCl→[AR-N2]+Cl-+H2O35
有的农药如对氨基苯磺酸钠,可用亚硝酸钠标准溶液直接滴定测定其含量,若是芳香族硝基取代时,如对硫磷、除草醚等,分子中的硝基可被冰醋酸—锌粉或醋酸、盐酸—锌粉等还原成氨基后,再用亚硝酸钠标准溶液滴定。在滴定中必须注意如下几个问题:(1)酸度:从反应式可看出,1克分子芳香胺需2克分子盐酸,实际测定时酸用量超过理论量,有时达3—4个克分子,因为在强酸介质个能加速反应,还能增加重氮盐的稳定性,当酸度不足时,生成的重氮化物与未被重氮化的芳香伯胺偶合,生成重氮氨基化合物造成测定结果偏低。[AR-N2]+Cl-+ARNH2→AR-N=N-AR+HCl酸量过多时会阻碍芳香伯胺的解离,减慢重氮化速度,重氮化反应多使用盐酸,因为在盐酸中比在硝酸、硫酸中反应快。(2)温度:在15—25℃进行重氮化滴定较合适。(3)滴定过程中应不断搅拌,因接近终点时溶液中伯胺量很少,要逐滴加入并充分搅拌。(4)溴化钾克加速反应:在测定对硫磷、除草醚时,芳香胺的对位取代基对重氮化反应速度有影响,在滴定前加入溴化钾,可发生下列反应:NOBr的解离常数较NOCl小、,能加快重氮化反应。(5)终点的判定,常用外指示剂法,以碘化钾淀粉为指示剂,滴定至终点时,由过量亚硝酸钠产生的亚硝酸将碘化钾氧化成碘,遇淀粉变蓝色。2NO2-+2I-+4H+→I2+2NO+2H2O将糊状淀粉指示剂倒在白瓷板上,铺成约1mm均匀薄层,在近终点时,用玻璃尖端沾取溶液少许,划过涂有碘化钾淀粉的白瓷板,即时出现蓝色条痕,在搅拌后,沾取溶液划过白瓷板仍显蓝色即已达终点。(五)非水滴定法弱酸性、弱碱性、在水中溶解度很小的农药难以在水溶液中进行酸碱滴定,采用各种非水溶剂作为滴定介质,从而扩大酸碱滴定范围,在水以外的溶剂中进行滴定分析称为非水滴定法。农药中含氮的杂环化合物很多,它们带有微弱的碱性或酸性,有的在水中溶解度小,有的在水溶液中滴定没有明显的突跃,难以掌握终点,采用有机溶剂或不含水的无机溶剂作为滴定介质,不仅能增加农药的溶解度而且对改变它们的酸碱性或其强度,使滴定反应能顺利进行。对于弱碱性化合物,用乙酸、丙酮、苯、三氯甲烷等溶解,以高氯酸标准溶液滴定。对于弱酸性化合物,用二乙胺、乙二胺和二甲基甲酰胺、苯、乙腈等溶解,以氢氧化四丁基铵标准溶液滴定。具有含氮杂环的农药,大多数可以进行非水滴定,如多菌灵分子结构为:,但具有弱酸性,在乙酸中可增强它的碱性,因此多菌灵样品经薄层色谱分离后,溶于三氯甲烷、乙酸酐后,加入冰醋酸、用高氯酸滴定。可测定其含量,我国多茵灵原药的国家标准中采用此方法(GB6697—86)。其它含氮农药如二嗪磷、蚜威和三唑酮等亦可用此法测定。35
二、电位滴定法和永停滴定法利用测定溶液的电极电位和浓度之间的关系来确定物质含量的方法称电位分析法。分析法包括直接电位法和电位滴定法。直接电位法是根据电极电位与离子浓度(活度)之间的对数关系,直接测出离子浓度的分析方法,如用酸度计测定溶液中的pH值等。电位滴定法是利用滴定过程中电位变化的突跃来确定终点的容量分析方法,是一种不用指示剂的滴定方法,根据溶液中电位的跃变来作为滴定终点的指示,所以终点的确定比一般容量法准确,客观,在有色溶液和浑浊溶液中均可滴定。在农药分析中常用于中和滴定、沉淀滴定和氧化还原滴定等。根据农药分析要求,本章还将介绍永停滴定法.(一)电位分析的几个基本概念1.原电池是借助于氧化还原反应得到电流的装置,以铜锌原电池为例(如图9—1)锌和硫酸锌溶液、铜和硫酸铜溶液各组成半电池,将锌片和铜片用导线联接,导线中串连一个电流表,同时将两溶液用含有琼胶的氯化钾饱和溶液的盐桥相联。锌失去电子发生氧化反应,电子由导线传到铜片上,Cu2+在铜片上得到电子发生还原反应析出金属铜。盐桥起通路作用,整个装置形成回路,放出的化学能变为电能,电流表指针发生偏转,锌极为负极,铜极为正极。2.电极电位原电池的两个电极产生的电流是由于两电极间存在电位差的缘故。当金属片插入它的盐溶液时,金属越活泼,溶液越稀,金属离子进入溶液中的趋势则愈大,金属表面留下电子而带负电,溶液中存在的金属离子带正电,在金属的周围就聚集成双电层,金属和溶液间便产生电位差;另一方面,溶液中的金属离子亦有从溶液沉积到金属表面的趋势,溶液愈浓,这种趋势也越大,也会形成双电层产生电位差。金属和溶液之间的电位差称为电极电位。组成原电池两电极的电极电位是不同的,两电极用导线连接即产生电源,该两电极的电极电位差称电动势。电极电位可由奈斯待(Nerst)方程式表示:Aox+ne=Ared(25℃)(9-1)E——平衡时的电极电位,E0——标准的电极电位;n——电极反应中参加反应的电子数目;[Aox]——氧化态活度;35
[Ared]——还原态活度。电极电位的绝对值是无法测量的,为了测量其相对值,早期用氢电极作标准电极,为负极,人为地规定其电极电位为零;待测电极作正极组成电池,此电池的电动势就是待测电极的电极电位。在25℃溶液中金属离子浓度为1mol/L时,所测得的电位值为该金属电极的标准电极电位,但实际测定时溶液的酸度、沉淀与络合物等都会有影响,标准电极电位与实际电位有很大差别,因此引入“克式量电位”。(二)参比电极与指示电极标准电极电位的测量是由标准氢电极和欲测电极组成电池测其电动势而得。标准氢电极的电位规定为零。表9-3银-氯化银电极的电极电位KCl溶液(mol/L)E(V)0.11.0饱和0.28800.22230.20001.参比电极电位值已知的电极称参比电极。标准电极的电位是已知的,但由于装配复杂,使用不便,已不常用。在实际工作中常用银-氯化银电极或甘汞电极作参比电极、溶液浓度或温度发生变化时,电极电位基本保持不变,且装置简单,使用寿命长。银-氯化银电极(图9-2)是由银丝上镀一薄层氯化银,浸于一定浓度的KCl溶液组成的:Ag│AgCl(固),KCl溶液,其电极电位间接决定于Cl-的活度[αCl-]。如银-氯化银电极的反应是:其电极电位决定于溶液中Ag+的活度[αAg+],可用下式表示:E[Ag]=E°[Ag+/Ag]+0.059lg[αAg+](9-2)而溶液中Ag+的活度决定于Cl—的活度,因为溶液中是难溶盐AgCl的饱和溶液,Ag+的活度由下列反应的平衡决定:其溶度积Ksp=[αAg+][αCl-][αAg+]=Ksp/[αCl-]以此式代入(9-2)式:E[Ag]=E°[Ag+/Ag]+0.059lgKsp-0.059lg[αCl-]前两项为常数,令E°’=E°[Ag+/Ag]+0.059lgKsp所以银—氯化银的电极电位为:E[Ag]=E°’-0.059lg[αCl-](9-3)即银—氯化银的电极电位决定于C1—活度,在固定C1—活度的条件下,可作为参比电极。在25℃时,不同浓度的KCl溶液的电极电位如表9-3。银-氯化银电极构造简单,体积小,电极电位不受温度变化的影响,广泛用作玻璃电极的内参比电极。甘汞电极的构造如图9—3所示。它具有两个玻璃套管,内套管封接一根铂丝,铂丝插入厚度为0.5—1.0cm35
的纯汞中,汞下装有甘汞、汞与少许氯化钾溶液研磨而成的糊状物,外套管装入KCl溶液。电极下端与被测溶液接触处熔接玻璃砂芯或陶瓷芯构成电极,其电极反应是:甘汞电极的电极电位在一定温度下亦决定于Cl—的活度,当电极内KCl溶液浓度一定时,电位值亦一定。其电极电位稳定、制备方便,经常用作参比电极。在25℃时浓度为0.1mol/L,1mol/L和饱和KCl溶液的甘汞电极,其标准电位分别为0.3370,0.2851,0.2443V。使用0.1mol/LKCl的电极温度系数最小,但制备不如饱和KCl溶液方便。2.指示电极能指示被测离子活度(或活度比)的电极称指示电极。指示电极应符合如下要求:①电极电位与被测溶液离子活度的对数值应成直线关系,即应符合奈斯特方程式;②对离子活度的变化响应快,再现性好;③使用方便,结构简单。在农药分析中常用的指示电极有活性金属电极,如银电极,是银丝浸在硝酸银溶液中构成;惰性金属电极如铂电极,它本身在溶液中并不参加反应,只是作为氧化还原反应交换电子的场所;测量pH值用的玻璃电极属于薄膜电极,将在下一节中讨论。(三)pH值的电位测定法用电位法可以测定溶液中的pH值,是以玻璃电极为指示电极,饱和甘汞电极为参比电极组成电池,玻璃电极为负极,甘汞电极为正极。装置如图9—4。1.玻璃电极玻璃电极是专用于测定pH值的指示电极。由一支玻璃管下端接一个特殊质料的球形薄膜制成,膜厚约30—l00μm、膜内装—定pH值的缓冲溶液和一定浓度的氯化钾,溶液中浸一根Ag—AgCl电极作内参比电极,当玻璃膜浸入水中,其表面即吸收小分,在膜内外形成两层溶胀的水化胶层。见图9—5所示。35
当玻璃电极浸入被测溶液中,玻璃膜处于H+活度一定的缓冲液和试液之间,膜内的水化胶层与缓冲溶液间产少相界电位E内,膜外水化胶层则与试液间产生相界电位E外,水化胶层内外侧的电位差称膜电位E膜。缓冲溶液中氢离子活度是一定的,为一常数,则(9-4)式中,K是玻璃电极的常数。(9—4)式说明,玻璃电极的膜电位与试液的pH成直线关系。2.测pH值的原理测定pH装置如图9-4,其电动势可表示位:以(9-4)式代入:由于电极与持测溶液之间的界面上有液体接界电位,玻璃膜内外产生不对称电位35
,则两极间的电动势为,,,对同一电极来说都是常数以表示即则:(9-5)这是电池电动势与待侧溶液pH值的关系式、测定电池电动势即可求出溶液的pH值。但是不对称电位和液接电位,在一定条件下,虽有定值却不易测量和计算。因此,总常数难以测定,通常采用两次测量法,即先用标准溶液进行校正,然后再测定待测溶液的pH值。3.标准缓冲溶液电位法测定pH值是以标准溶液作基准的。因此,标准缓冲溶液的准确配制十分重要。方法如下:(1)0.05mol/L邻苯二甲酸氢钾溶液:称取110℃干燥2—3h的邻苯二甲酸氢钾10.12g溶于蒸馏水中,稀释至1L。(2)0.025mol/L磷酸二氢钾和0.025mol/L磷酸氢二钠混合溶液:分别称取120℃烘干2—3h的磷酸二氢钾3.39g和磷酸氢二钠3.53g于烧杯中,加蒸馏水溶解,稀释至lL.干燥湿度切忌过高,否则会产生缩合磷酸盐。(3)0.01mol/L硼砂溶液:称取硼砂(Na2B4O7·10H2O)3.80g溶于蒸馏水中(硼砂不要烘),稀释至1L。该溶液易吸收CO2改变pH值,故配制溶液的蒸馏水应预先煮沸并冷却,同时瓶口应装置碱石棉管。(4)0.05mol/L四草酸氢钾溶液:称取在54℃烘干4—5h的四草酸氢钾[KHC2O4·H2C2O4·2H2O]12.61g溶于蒸馏水中,稀释至lL。(5)25℃饱和酒石酸氢钾溶液:在磨口玻璃瓶中装入蒸馏水和酒石酸氢钾(约20g/L),剧烈摇荡使其充分溶解,使用前在22—28℃将清液倾出,为防止溶液发霉,溶液中可加入0.9g/L百里酚。(6)饱和氢氧化钙溶液;在磨口玻璃瓶或聚乙烯塑料瓶中,加入蒸馏水和过量氢氧化钙(约10g/L),温度控制在25℃,剧烈振动30min,使用时倾出清液。以上6种溶液系国家标准局颁发的pH基准缓冲溶液,其不同温度下的pH值列于表9—4。温度℃0.05mol/L四草酸氢钾饱和酒石酸氢钾0.05mol/L邻苯二甲酸氢钾0.025mol/L磷酸二氢钾与0.025mol/L磷酸氢二钾0.01mol/L硼砂饱和氢氧化钙101520251.671.671.681.68---3.564.004.004.004.006.926.906.886.869.339.289.239.1813.0112.8212.6412.4635
30354045501.681.691.691.701.713.553.553.553.553.564.014.024.034.044.066.856.846.846.836.839.149.109.079.049.0212.2912.1311.9811.8311.70(四)电位滴定法它是一种用电位法测定滴定终点的方法。与直接电位法不同,不是由电极电位的数值来计算待测离子浓度,而是以测量电位的变化为基础的。电位滴定装置如图9—6,是由指示电极.参比电极浸于被测溶液中组成电池,在电磁搅拌器不断搅拌下从滴定管缓缓滴入已知浓度的标准试剂,溶液内发生化学反应,被测离子的浓度发生变化,指示电极的电位也相应发生变化,到达等当点附近,电位变化较大,产生突跃;等当点后,电位变化较小,根据电极电位变化的突跃可判断等当点。电位变化突跃不如指示剂直观,在等当点附近要求每滴加0.1ml测量一次电位,通过作图法来确定终点。1.终点确定(1)E—V曲线法:以加入滴定剂的毫升数(V)为横坐标,电位计读数(E)为纵坐标,绘制如图9—7(a)所示的滴定曲线。作两条与滴定曲线相切45°的直线,两切线间的平行等分线与滴定曲线的交点即为滴定终点。以0.1mol/LAgNO3溶液滴定NaCl溶液35
为例,银电极为指示电极,饱和甘汞电极为参比电极,滴定过程的电位值如表9—5。(2)一级微商法:即ΔE/ΔV—曲线法,当终点电位突跃变化不明显,用E—V曲线法不易确定终点时,可采用此法。是以每单位滴定体积的改变所引起的电位改变(ΔE/ΔV)对相邻滴定体积的平均值(7)作图,即根据表9—5数据以ΔE/ΔV作纵坐标,以r为横坐标绘制图9—7(b).曲线最高点所对应的电位为终点电位,虚线指示滴定终点的体积。从表9—5可见,在24.30一24.40m1之间,电位的增量为830mv,为最大突跃,因此终点体积约为24.35ml。(3)二级微商法:即/曲线法,为了更准确地确定终点体积,可采用二级微商法,,为相邻的一次微商之差除以ΔV,根据表9—5的数据一极微商最大,二级微商等于零时,所对应的体积为滴定终点。即在24.30一24.40ml之间,必要时,可用内质法计算其准确终点体积。滴定达到终点时,消耗滴定剂的体积为24.34ml。2.农药分析中常用的电位滴定类型(1)中和滴定:一般选用饱和甘汞电极作参比电极,玻璃电极作指示电极、用pH计测定溶液的pH值作纵坐标,滴定体积作横坐标绘出曲线或上述二次微商法确定终点,前述容量法中可以使用酸碱滴定来测定其含量的农药,都可使用电位滴定法。氨基甲酸酯农药如速灭威、仲丁威、异丙威、甲萘威、涕灭威和克百威等,采用薄层—定胺法时,最后以盐酸标准溶液滴定水解生成的甲胺,接近终点时,每次准确加入0.1ml、用二次微商法求出终点。(2)沉淀滴定:在农药分析中常用银量法,是以银电极为指示电极.饱和甘汞电极作参35
比电极,以硝酸银作标准溶液,可测Cl-、Br-、I-、CNS-等离子。由于其它氯离子有干扰,甘汞电极不能直接插在待测溶液中。用硝酸钾盐桥将待测溶液与甘汞电极分开、根据滴定的电位跃迁确定等当点,计算出待测溶液的浓度。在第一节中讨论的可用容量法进行沉淀滴定的农药均可采用电位法。如敌敌畏样本经薄层板分离及碱解后、将有效成分溶于水及乙醇中.加1+3硝酸中和至甲基橙转红,并过量2—3滴,插入银—甘汞电极对,接电位计、在搅拌下用0.0lmol/L硝酸银标准溶液滴定,接近终点时,每次准确滴入0.1ml。以二次微商法求出终点,如氯菊酯用氨氧化钾—乙二醇溶液水解后,先用1+3硝酸中和至甲基橙变红并过量2—3滴,插入银—甘汞电极对,接电位计。用0.0lmol/L硝酸银标准溶液滴定,以二次微商法求终点,在测定溴氰菊酯,氯氰菊酯,氰戊菊酯及百菌清等农药时,电位滴定条件与氯菊酯相同,但在加硝酸中和后,必须加5ml甲醛以除去一CN基的干扰,否则结果偏高。(3)氧化还原滴定:在第一节容量分析法可进行氧化还原滴定的农药,都可以用电位滴定法完成。一般用铂电极作指示电极,甘汞电极作参比电极。为了响应灵敏,铂电极的表面必须洁净光亮。(4)非水滴定法:可进行非水滴定的农药如二嗪磷、抗蚜威、三唑酮等经薄层分离后,用氯仿和乙酸酐将有效成分洗入50m1干燥烧杯中,加5ml冰乙酸,l滴结晶紫指示剂,以玻璃电极为指示电极和甘汞电极为参比电极,甘汞电极的夹层中放1mol/L乙酸钠的冰醋酸溶液,接通精密电位计,用高氯酸标准溶液滴定、临近终点结晶紫由紫变蓝色时,每次加0.1ml标准溶液并记录电位变化,用二次微商法求出终点。(五)永停滴定法它又称死停滴定法,是将两支相同的铂电极插入待测溶液中,在两个电极间另加一小电压(10一200mV),然后进行滴定。观察滴定过程中两个电极间的电流变化,根据电流变化情况确定滴定终点,所以此法属于电流滴定范畴。永停滴定法装置简单,准确度高,容易确定终点,是重氮化法和卡尔·费休(KarlFisher)水分测定法确定终点的法定方法。当溶液中同时存在氧化还原电对,如I2与I-在溶液中同时插入相同的两支铂电极时,由于电极电位相等,其电动势等于零。若在两电极间外加一小电压,则接正端的铂电极将发生氧化反应;。接负端的铂电极上将发生还原反应:。溶液中发生电解反应,并有电流通过。这种外加很小电压就能产生电解反应的电对称可逆电对。若溶液中的电对是S4O62-/S2O32-时,阳极可以发生还原反应:2S2O32-→S4O62-+2e,而阴极不能发生氧化反应,这种电对称为不可逆电对,外加很小的电压不能产生电解反应。不同电对间反应不同,有三种不同的电流变化情况,其终点判断亦不同。在农药分析中主要使用以下两种:(1)滴定剂是可逆电对,被测物是不可逆电对,如用碘滴定硫代硫酸钠。I2+2S2O32-→2I-+S4O62-等当点前,溶液中只有不可逆的S4O62-/2S2O32-电对,它不发生电解反应,电极间没有电流通过,当到达等当点并过量半滴I2后,溶液中存在I2/2I-可逆电对,电极上即发生电解反应,溶液有电流通过,即可指示终点到达。等当点后,I2不断增加,电流不断增大,如图9—8(a)。(2)滴定剂为不可逆电对,被测物为可逆电对,如硫代硫酸钠滴定碘。等当点前溶液中存在I2/2I-电对,有电流通过但逐渐减小,当滴定至等当点过量一点时,I2则全部变I-,没有可逆电对,无电解发生,电流降到最低点,指示终点到达,如图9—8(b)。35
在上面讨论的亚硝酸钠滴定法,可以采用永停法确定终点,比使用内外指示剂都准确方便。如用NaNO2标准溶液滴定,一NO2基被还原成一NH2基的对硫磷;终点前溶液中不存在可逆电对,电流计指针停止在零位不动,达到终点并稍有过量NaNO2,则溶液中HNO2及其分解产物NO作为可逆电对同时存在,两个电极上起如下的电解反应:阳极:NO+H2O→HNO2+H++e阴极:HNO2+H++e→NO+H2O电路中有电流通过,电流计显示偏转并不再回到零位。在进行卡尔—费休法测定微量水分时,采用永停法指示终点,比用碘作为自身指示剂更加准确方便,样品中的水与卡尔—费休滴定剂起如下反应,终点前溶液中不存在可逆电对,电流指针停止在零位不动。达到终点并稍有过量的I2,则溶液中便有I2及I-可逆电对同时存在两电极上起如下反应;阳极,2I-→I2+2e阴极:I2+2e→2I-电路中有电流通过,电流计指针显示偏转并不再回到零位。永停滴定法的仪器装置如图9—9所示。B为1.5V干电池,R和R1为变阻器,R的电阻约5000Ω,Rl的电阻约500Ω,G为电流计,R2为电流计的分流电阻,可调节电流计的灵敏度,E’、E是两个完全相同的铂电极。滴定开始时,先用Rl调节适当的外加电压(约几毫伏至几十毫伏),使溶液中通过电流,滴定时不断记录电流表数值,绘制滴定曲线确定终点。35
(六)电位滴定分析农药时电极的选择电位滴定分析农药时电极的选择可参考表9-6。35'
您可能关注的文档
- 校园社区网创业计划书.doc
- 校园网络组建方案计划书.doc
- 校园自助式投币洗衣机创业计划书.doc
- 校园西式快餐店创业计划书.doc
- 校园超市创业计划书.doc
- 校园零食小铺创业计划书.doc
- 校园餐饮项目创业计划书.doc
- 校园驿站创业计划书.doc
- 核桃种植可持续性精准扶贫策划方案.docx
- 格兰仕网站架构方案书.doc
- 格力旺角商业文化中心销售策划方案.doc
- 格升软件有限公司创业计划书.doc
- 格子铺项目创业计划书.doc
- 桂林元气罗汉果创意有限公司创业计划书.doc
- 桂林工学院学生社团办公楼组织设计.doc
- 框架设备间施工方案.doc
- 桌上游戏创业计划书.doc
- 桌游工作室创业计划书.doc
相关文档
- 施工规范CECS140-2002给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程
- 施工规范CECS141-2002给水排水工程埋地钢管管道结构设计规程
- 施工规范CECS142-2002给水排水工程埋地铸铁管管道结构设计规程
- 施工规范CECS143-2002给水排水工程埋地预制混凝土圆形管管道结构设计规程
- 施工规范CECS145-2002给水排水工程埋地矩形管管道结构设计规程
- 施工规范CECS190-2005给水排水工程埋地玻璃纤维增强塑料夹砂管管道结构设计规程
- cecs 140:2002 给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程(含条文说明)
- cecs 141:2002 给水排水工程埋地钢管管道结构设计规程 条文说明
- cecs 140:2002 给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程 条文说明
- cecs 142:2002 给水排水工程埋地铸铁管管道结构设计规程 条文说明