

- 4.75 MB
- 2022-04-22 11:47:53 发布

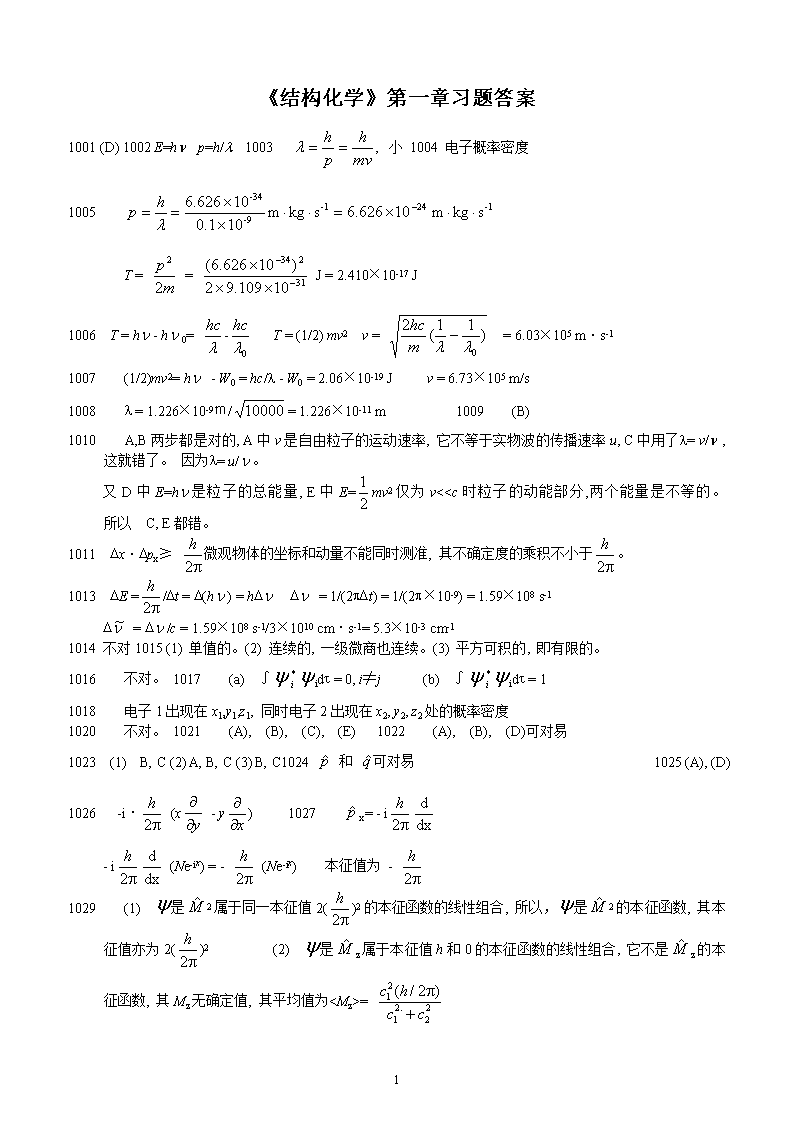
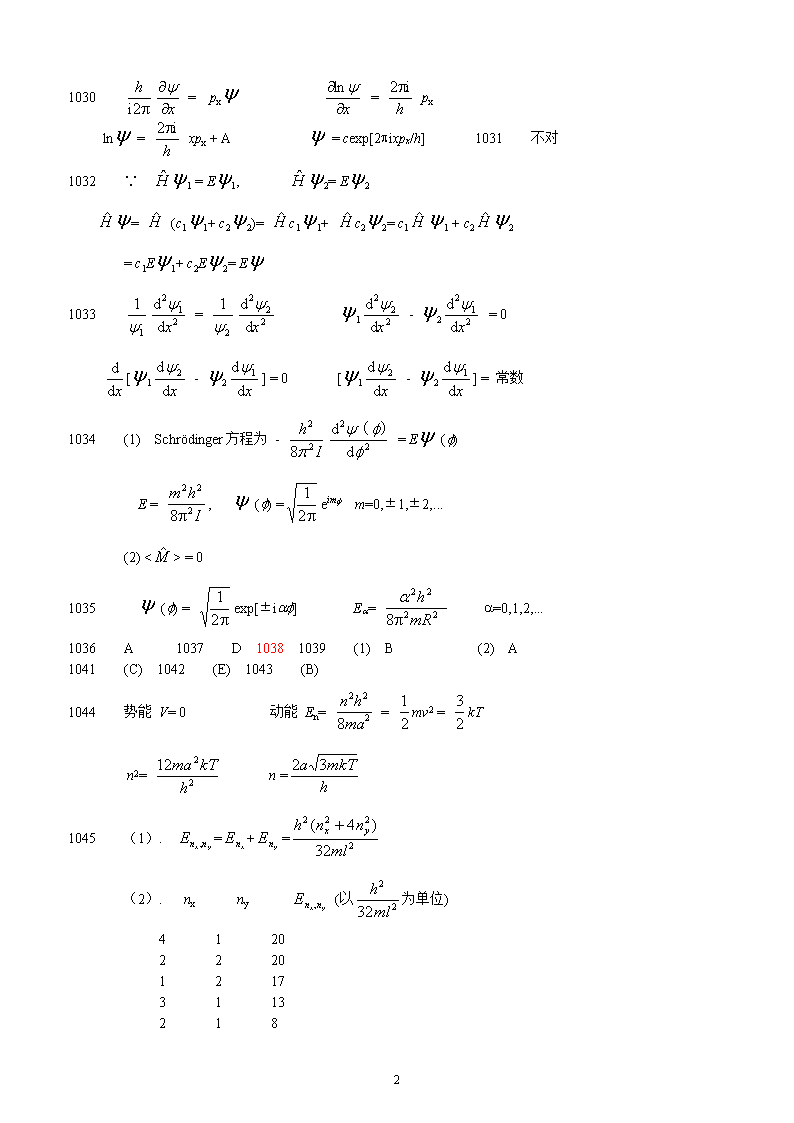
- 1、本文档共5页,可阅读全部内容。
- 2、本文档内容版权归属内容提供方,所产生的收益全部归内容提供方所有。如果您对本文有版权争议,可选择认领,认领后既往收益都归您。
- 3、本文档由用户上传,本站不保证质量和数量令人满意,可能有诸多瑕疵,付费之前,请仔细先通过免费阅读内容等途径辨别内容交易风险。如存在严重挂羊头卖狗肉之情形,可联系本站下载客服投诉处理。
- 文档侵权举报电话:19940600175。
'目录第一章答案----------------------------------------------------------------------------1第二章答案---------------------------------------------------------------------------26第三章答案---------------------------------------------------------------------------47第四章答案---------------------------------------------------------------------------63第五章答案---------------------------------------------------------------------------7183
《结构化学》第一章习题答案1001(D)1002E=hp=h/l1003小1004电子概率密度1005T==J=2.410×10-17J1006T=h-h0=-T=(1/2)mv2v==6.03×105m·s-11007(1/2)mv2=h-W0=hc/l-W0=2.06×10-19Jv=6.73×105m/s1008l=1.226×10-9m/=1.226×10-11m1009(B)1010A,B两步都是对的,A中v是自由粒子的运动速率,它不等于实物波的传播速率u,C中用了l=v/,这就错了。因为l=u/。又D中E=h是粒子的总能量,E中E=mv2仅为v<=83
1030=px=pxln=xpx+A=cexp[2pixpx/h]1031不对1032∵1=E1,2=E2=(c11+c22)=c11+c22=c11+c22=c1E1+c2E2=E1033=1-2=0[1-2]=0[1-2]=常数1034(1)Schrödinger方程为-=E(f)E=,(f)=eimfm=0,±1,±2,...(2)<>=01035(f)=exp[±iaf]Ea=a=0,1,2,...1036A1037D10381039(1)B(2)A1041(C)1042(E)1043(B)1044势能V=0动能En==mv2=kTn2=n=1045(1).=+=(2).nxny(以为单位)412022201217311321883
1151046(1)=sinn=1,2,3,…(2)E=;(3)1/2(4)增长(5)=sinsinE=+1047(1)211(x,y,z)=sinxsinysinz(2)(a/4,a/2,a/2)(3a/4,a/2,a/2)(3)610483,41049(非)1050E=共有17个状态,这些状态分属6个能级。1051=-+x2=E=E=h1052到5所需能量为最低激发能。1053P=sin2()dx=0.5+=0.8181054一维势箱E1==6.03×10-8J静电势能V=-=-2.3×10-13J由于动能大于势能,体系总能量大于零,不能稳定存在。发出h≈E1的射线(b射线)。1055库仑吸引势能大大地小于电子的动能,这意味着仅靠库仑力是无法将电子与质子结合成为中子的,这个模型是不正确的。1056DE=[(22+22)-(12+22)〕=l===86.2nm1059(1).该函数是一维箱中粒子的一种可能状态,因sin及sin是方程的解,其任意线性组合也是体系可能存在的状态。(2).其能量没有确定值,因该状态函数不是能量算符的本征函数。(3).=83
1060(1)n=sinP1/4=∫dx=-sin(2)n=3,P1/4,max=+(3)P1/4=(-sin)=(4)(3)说明随着粒子能量的增加,粒子在箱内的分布趋于平均化。1061Y111(x,y,z)概率密度最大处的坐标为x=a/2,y=b/2,z=c/2Y321(x,y,z)状态概率密度最大处的坐标为:(a/6,b/4,c/2),(a/6,3b/4,c/2),(a/2,b/4,c/2),(a/2,3b/4,c/2),(5a/6,b/4,c/2),(5a/6,3b/4,c/2)1062是;=+=+=1063要使波能稳定存在,其波长l必须满足驻波条件:n=l,n=1,2,…考虑到德布罗意关系式,从上式可得:p==在一维势箱中,势能V(x)=0,粒子的能量就是动能E==1064(1)2(2)3(3)41065Dl=l2-l3=-=a-a=a1066一维势箱En=DE=E2-E1=-=l==对电子l=11.00nm对a粒子l=8.07×104nm106721068(1)[-+kx2]=E(2)E===h(3)x=0时,=0,有最大值0(0)=()1/4最大值处x=002=()1/2=1069已知势箱长度之比为300pm:100pm=3:1假设==4eVh2/(8m)=432eV83
EH=[·]3=4×32×3=108eV1070=cosxE=,n=1,3,5,…=sinxE=,n=2,4,6,…1071(1)l=2×10-10m(2)l=1.1×10-8m(3)T=5.43×10-17J1072(1).E=(2).(3).(4).=01073当时,107410751076以作用于不等于常数乘,即可证得。可和交换.1077同理=b/2=c/2所以,粒子的平均位置为(a/2,b/2,c/2)1078一维箱长l=(k-1)a,En=k=偶数,k=奇数,83
1079E=为使平方可积,取1080T==1.016×10-17J1101(C),(D)1102(A)1103(1)(2)(3)(4)1104光子波长自由电子的波长质量为300g的小球的波长1105(589.0nm)(589.6nm)1106(1)83
(2)不能1107(1)(2)可以(3)1108(B)1109中子:1110不能观测到波动性能观测到波动性能观测到波动性加速后不能观测到波动性1111不能1112(1)(2)(3)(4)(5)111383
11151114(C)1116(C)1117或1118(A),(B)1119(D)??E=L=8Rc-c=1120pm所以最低激发能为DE=E5-E4==4.323×10-19J=2.698eVDE=h=hc/ll==459.8nm460nm为蓝光,即该分子吸收蓝色光。在白光中表现为红色。11211122观察到的最低跃迁频率对应于n=1向n=2的跃迁。故箱子长度为8.27×10-10m1123势箱中故=2l/n=(200/n)nmn=1P1=0.0400n=2P2=0.0001n=1时无节面,概率密度最大在50nm处。n=2时节面数=n-1=1,节面在50nm处,概率密度最大在25nm和75nm处。1124En=n2h2/(8ma2)n=1,2,3,...83
1125立方势箱的能量表达式为nxnynzE(以为单位)g(简并度)111311211126321112222193212113131113311222121123132321146312231321第四个能级能量为(11h2)/(8ml2),简并度为3。第六个能级能量为(7h2)/(4ml2),简并度为6。1126估算的吸收光的波长506.4nm与实验值相接近。1127l=1.05nm1128(A).不是(B).是,本征值为n2h2/(4l2)(C).不是(D).是,本征值为n2h2/(8ml2)1129将代入方程说明是方程的解。将代入方程说明也是方程的解。83
1130`故x+iy是本征函数,本征值为故x-iy是本征函数,本征值为故z是本征函数,本征值为01131n=1,2,3,…节面愈多,波长愈短,频率愈高,能量亦愈高。1132是量子化的,因为对振动x≈1×10-9cm,而p=mv=[(2×10-3)/(6.02×1023)]×103=J·m-1·s-1由测不准关系,p≈h/x=(6.626×)/(1×10-11)=6.626×10-23J·m-1·s-1p~p,所以测不准原理起作用,能量是量子化的.对转动x≈1×10-8cm=1×10-10m=0.1×p=3.32×J·m-1·s-1=6.626×J·m-1·s-1转动能量也是量子化的。1133丁二烯吸收发生在紫外区,所以是无色的,维生素A吸收在可见部分的高能区见到绿和红的混合。1134x=3.883×10-10m,与荧光屏电子显像管大小相比,可忽略。1135太阳能发电机每小时每平方米从太阳获得最大能量为3×105J。电站需要采光面积为6×107m2。1136不能。83
1137每秒发射5.33×个光子.要5分35秒(335秒)。1138电子能量:T=1.01×10-17J中子能量:T=5.54×J1139≈11nm在X-射线范围。1140第一吸收带是由HOMO到LUMO跃迁产生。对本题HOMOk=n;LUMOk=n+1;所以即114121201142A=B=1/时,最大。A=-B=1/时,最小。1144(1)k1=(2/h)(2mE)1/2,k2=(2/h)[2m(V-E)]1/2,(2)=[(V-E)/E]1/2=1,(2)对于此电子迁移,隧道效应是主要的。1145E=E0+1/211461147设u和v是两个任意函数,由此得证1149设u1,u2,...,,...是算符的分别属于本征值,...的本征函数,则有83
可得根据的厄米性,从上式可得1150按厄米算符的定义,有同时下列本征方程成立:代入上式,得:由此可得故必为实数。1151设:(1).和是的本征函数,相应的本征值为E1和E2。(2).证:只有当E1=E2时,才有即,才是原算符的本征函数。1152115383
由此得证1154(1).可以;(2).可以;(3).不可以(4).可以1155(1).∫u*()vd=∫u*vd51+∫u*vd=∫(u)*vd+∫(u)*vd=∫[(u)*+(u)*]vd=∫[(u)+(u)]*vd=∫[()]*vd由此得证(2).∫u*v=∫u*(v)=∫(u)*(v)=∫(u)*v=∫(u)*v=∫(u)*v由此得证1156可用数学归纳法证。1157(1)17(2)51158(1)=171233cm-1(2)E=171233/8065=21.23(eV)E=21.23×1.60×10-19=3.40×10-18(J)(3)Ek=21.23-15.759=5.47(eV)1159=(h/)/E=(h/)/(h)=1/()=1/()(1)=0.1cm-1=1/()=1/(×3××0.1)=5.3×(s)(2)=1cm-1=1/()=1/(×3××1)=5.3×(s)(3)=100MHz=100106s-1=1/()=1/(×100×)=1.5×(s)1160=1120(pm)1161是共同的本征函数为和的线性组合,是共同的本征函数83
是共同的本征函数1162不正确,微观体系力学量是量子化的。1163m===0.05kg1164P==n=1,P=n=2,P=.n=2时,粒子出现在0—a/4区间概率更大些。1165====12是,本征值为12。1166E/h2/8ml29nx=2ny=2nz=1;nx=1ny=2nz=2;nx=2ny=1nz=26nx=1ny=1nz=2;nx=2ny=1nz=1;nx=1ny=2nz=13nx=1ny=1nz=11167E/h2/8ml283
10nx=3ny=1;nx=1ny=38nx=2ny=25nx=2ny=1;nx=1ny=22nx=1ny=11168归一化条件:A2(,a是归一化的。B,b不是归一化的。归一化因子即。1169======归一化波函数11702归一化因子N=,归一化波函数=。1171==归一化波函数。117283
==i2=0(被积函数为奇函数)=01173===0(被积函数为奇函数)1174=====01175==2====2=归一化波函数1176====归一化因子归一化波函数83
1177===,归一化因子归一化波函数11781179设为宇称算符,其本征函数和本征值分别为G(x,y,z)和g。则本征方程为G(x,y,z)=gG(x,y,z)G(x,y,z)=G(x,y,z)=gG(x,y,z)=gG(x,y,z)=g2G(x,y,z)g2==1g=1即G(x,y,z)=G(x,y,z)即G(-x,-y,-z)=G(x,y,z)若g=1,则G(-x,-y,-z)=G(x,y,z),即G(x,y,z)为偶函数。若g=-1,则G(-x,-y,-z)=-G(x,y,z),即G(x,y,z)为奇函数。宇称算符的本征函数非奇即偶。1180一个自由电子具有多重基态。多重性为2,即双重态。a.一维势箱中的8个电子排布如图,只可能是单重态。E/h2/8ml216n=49n=34n=21n=1118183
==2.381182eV1=h1-W0……………(1)eV2=h2-W0……………(2)(2)-(1),得eV2-eV1=h2-h1,e(V1-V2)=h(2-1)则=J·s=J·s1183根据求平均值的公式,,ci为第i项前的组合系数。则=1184(1)爱因斯坦(2)德布罗意(3)海森堡(4)薛定谔(5)玻恩1185==是,本征值为-2。1186(1)(2)83
1187(1)(当n=2时)==(2)0a/4a/2ax1188E/18nx=3ny=113nx=2ny=110nx=1ny=11189根据n=30a/3ax当n=3时,粒子出现在区域中的概率为1/3。1190M2有确定值,因为L=1,所以M2=2。Mz无确定值,其平均值为:。1191由cH2:cLi2=9:1,cH2+cLi2=1,可得cH=0.949,cLi=0.336.83
1192===1193=0.01%´300m·s-1速度不确定度为原运动速度的0.01%.1194三维空间中自由粒子薛定谔方程因与r,q,f无关,故从而E=0因角动量M2是与q,f有关的一个算符,而是常数,故M2=0。1195P====区间,区间,区间1196==当n=1,当n=2,当n=3,1197P=====n=1,P=0.6089n=2,P=0.1955n=3,P=1/3.83
1198P===区间,区间,区间1199当与无关,==2角动量大小为。1200三维空间自由粒子的薛定谔方程当r为常数,与r,无关。==当与无关,1201三维空间自由粒子的薛定谔方程当r为常数,与r无关,83
==(+)=式中=1N=,1202=(+)=式中=1N=,为一常数,证毕。1203因未归一化,120483
=只有sin2x-cos2x是的本征函数,本征值是-4。1205只有sinxcosx是的本征函数,本征值是-4。1206该多烯中有6个电子,最高填充能级n=3,最低空能级n=4。=120783
1208120912101:1:11211=121212131214121583
1216式中=为常数,于是:1217三维空间自由粒子的薛定谔方程当与r无关,=(6+)==-983
N,12181219则=546:11220f(x)是的本征函数,本征值为。12211222《结构化学》第二章习题答案2001式中:r=(x2+y2+z2)1/22002(a)-13.6eV;(b)0;(c)0;(d)2,0,0;(e)083
2003(1)r=a0/3,(2)=a0/2,(3)2005(a)0(b)0(c)2.618a02006不对。2007不对。200822009(a)n,l(b)l,m(c)m2010(D)2011(C)根据函数的单值性可确定│m│的取值为0,1,2,...,但不能确定其最大取值l,│m│的最大值是由方程求解确定的。2012不对。2013不对。2014否。2015否。2016n=3,l=1,m=0。2017根据正交归一化条件2018(1)(-1/4)×13.6=-3.4eV(2)(3)90°2019将波函数与H原子一般波函数比较可得:n=3,l=2,E=(-1/9)×13.6eV=-1.51eV该波函数为实函数,无确定值,求平均值如下:20202021(1)(2)能量相同202283
为确定的常数,则复函数是算符的本征函数。按相似方法进行运算,对实函数得不到常数乘原函数,故不是的本征函数。2023<1/r>=1/a0=-e2/a0E=T+V=-e2/2a0=e2/2a02024证:因为s态波函数仅为半径r的函数,2025考虑到波函数的正交性和归一化可得R为里德堡常数(13.6eV)2026在x轴和y轴均无确定值,其平均值均为020272028l:0,1,2,3m:0,±1,±2,±3ms:±1/2总的可能状态数:2(1+3+5+7)=32种2029玻尔模型:,能量是由此推算而得,量子力学:M=0,能量由解薛定谔方程得到。2030(a)(b)出现在的概率为1(c)2031(a)(b)c12+c22(c)(d)1(e)(f)02032(a)A,B,C(b)A,B,C(c)A,C20331s,2s,3s,2pz,3pz,383
2034(a)-1.511(b)r及(c)能量以及角动量大小2035(a)-1.51eV(b)(c)66°2036(D)2037(A)2038(A)2039(C)2040不对,l确定后,轨道角动量的大小是能确定的,但其方向不能确定。2041是。2042不对。m相同的轨道,l值不一定相同,所以角动量不一定相等.2043径向部分有一个节面,其方程是r=120a0/Z,角度部分x2-y2=0得x=±y,得角度部分有两个节面,其方程分别是x=y;x=-y共有3个节面,把空间分成8个部分.2044相对概率是=sin290°/sin245°=2概率之比是2。20452046=0.32322047电子云极大值位置即极值位置,根据所以,电子云极大值在z轴上,距核为2a0处.2048平均来说,2p电子离核比2s电子要近。2049(1)0.764a0,5.236a0(2)0,4a0(3)2a083
2050a.u.2052(1)0(2)a0/2(3)a0/3(4)相等(5)122.4eV2053参看《结构化学基础》(周公度编著)p.582054参看《结构化学基础》(周公度编著)p.582055参看《结构化学基础》(周公度编著)p.582056参看《结构化学基础》(周公度编著)p.542058(1)1个节面,位置在通过坐标原点的xoy面上,平面形。(2)在z轴上,距原点2a0处。(3)略2059(a)根据径向部分节面数定义:n-l–1,则为0(b)角度部分节面数为l,即2(a)-3.4eV(b)电子云(c)或与成正比2062(a)(b)2063(a)核附近(b)离核a0处2064(a)一样(b)不一样2065(a)2(b)-1.51eV(c)(6)1/2h/(d)65.902066(a)3(b)1(c)02067(D)2068(D)2069(C)2070(C)2071(B)2072(D)2073(D)2074全部为(非)。2075不对。2076不对。2077不对。2078(1)(2)由2079He原子薛定谔方程为中心力场模型把原子核和两个电子所形成的势场看作是个中心力场,只是离核距离的函数。当用光激发时,根据跃迁选律:△S=0,△L=±1。其最低激发态为1s12p1,该状态的轨道角动量│M│=[l(l+1)]1/2=2080基态He原子的Slater行列式波函数为He原子第一激发态的Slater行列式波函数为83
208120822083(a)-13.6eV(b)-3.4eV(c)-4.5eV(d)-13.6eV2084E1>E2>E32086(A)2087(A)2088(C)2089(非)2090(是)2091(1)(2)(3)(4)=2[1s(1)]2,由于[1s]2是球对称的,所以氦原子基态电子云是球对称的。2092(a)(2L+1)(2S+1)(b)52093(D)2094(D)2095非2096(C)2097(B)组态全部光谱项为1D,3D(B)中不含3F4支项,因此是(A)排布。2098(A)2099(D)2100(C)2101V(1s22s22p63s23p64s23d3)4F3/221022P3/22103Z24294441基组态4s13d54s13d105s14d75s14d4基谱项7S2S5F6D能级最低的7S32S1/25F56D1/2光谱支项2104(a)(b)(c)(d)52105Ti[Ar]4s23d23F22106(1)1S0(2)3P0(3)3P2(4)2P3/2(5)4F3/22107S:3P2V:4F3/22108(1)2S1/2(2)2P3/2,2P1/2(3)2D5/2,2D3/2(4)2P3/2,2P1/22109Fe(1s)2(2s)2(2p)6(3s)2(3p)6(4s)2(3d)65D42110Co3+:5D4Ni3+:4F9/221112p23p1光谱项为4D,4P,4S,2F,2D(2)`!`,2P(3),2S光谱支项4D7/2,5/2,3/2,1/2;4P5/2,3/2,1/2;4S3/2;2F7/2,5/2;2D5/2,3/2(2)2P3/2,1/2(3)2S1/2括号中的数字表示该谱项重复出现的次数。21124G,4F(2),4D(3),4P(2),4S,2G(2),2F(4),2D(6),2P(4),2S(2),括号中的数字表示该谱项重复出现的次数。2113Li1s22s1光谱支项2S1/21s22p12P3/2,2P1/2Li2+2s1光谱支项2S1/22p12P3/2,2P1/283
Li原子是多电子原子,原子轨道的能级与n,l有关,所以组态1s22s1与1s22p1能量不等。Li2+是类氢离子,仅有一个电子,能级只与n有关,所以这两组态能量相等。2114能级由高到低次序为:1S01D23P23P13P0微观能态数155312115考虑到旋轨偶合,引出量子数J,光谱项分裂成光谱支项3P:3P2,3P1,3P0分裂成3个能级1P:1P1不分裂1D:1D2不分裂6S:6S5/2不分裂.21162P:光谱支项为2P3/2,2P1/2,其状态数分别为4和20。3P:光谱支项为3P2,3P1,3P0,其状态数分别为5,3,1。3D:光谱支项为3D3,3D2,3D1,其状态数分别为7,5,3。2D:光谱支项为2D5/2,2D3/2,其状态数分别为6,4。1D:光谱支项为1D2,其状态数为5。21172P3/2分裂为4个。2P1/2分裂为2个。2118旋轨偶合能级有3P2,3P1,3P0;施加外磁场上述能级进一步分别分裂为5,3,1个。2119pd组态的光谱项为3F,1F,3D,1D,3P,1Pp2组态光谱项为3P,1D,1S选择定则△S=0△L=0,±1所以允许的跃迁是3P→3P,3D1D→1F,1D,1P1S→1P21202p4和2p2相似,参看《结构化学基础》(周公度编著)p.84能量最低的光谱支项3P2(不是2p2的3P0)21212P3/2→2S1/2,2P1/2→2S1/2参看《结构化学基础》(周公度编著)p.812123En=-R/n2巴尔麦系m=2n=3对应最小,波长最长2124En=-R/n2巴尔麦系m=2对应波长最短,则最大之n=83
2125不能。赖曼系在紫外区,巴尔麦系在可见光区,帕邢系在红外区。2126此波长所对应的能量:E=1239.8/91.1eV=13.609eV是氢原子的电离能。2127(1)1;(2)9;(3)25.2128n=1,v0=2.19×106m·s-1n=10,v=2.19×105m·s-12129CrZ=24对K层n1=1Zk*=24L层n2=2ZL*=22和实验值228.5pm比较相差14%,主要是内层电子速度快,应进行相对论校正;有效核电荷数Z*的计算也是近似的。2130(a),0(b),0(c)45°,90°,135°213154.4eV2132He+的电离能为:13.6×Z2/n2=13.6×4eV=54.4eVHe的I1=[2×13.6×(2-0.3)2-54.4]eV=24.2eV21335.746eV2134I1=E(He+)-E(He)E(He)=E(He+)-I1=-78.98eV2135[-79.0-2×(-54.4)]eV=29.8eV213621372138(C)2139(1)180°,70.5°(2)45°,90°,135°2140(1)70.53°,180°(2)45°,90°,135°21412S,2S1/283
2143共有2个节面=54044'和=125016'这两个节面把空间分成3个部分。2144轨道角动量径向分布节面数角度部分节面数n-l-1l1s0002p013d022145△E×△t=h2146(a)自旋-轨道,(b)反对称的2147n,l,m,ms2148L,S,J,MJ2149a0/4.2150a021512152a0E0=-R,与真实能量一致。2154Pauling标度:已知xF=4.0,xA-xB=0.102△1/2△为A—B键的键能与A—A键和B—B键键能的几何平均值的差值。Mulliken标度:x=0.18(I1+Y)0.18为拟合常数2155(B)2156(B)2157根据量子数的限制,可得如下周期表形式12345678910111213141516满壳层者为惰性元素,即1,4,9,16等号元素。2158(D)2159(C)2160(B)2161(B)2162(1)r2R2dr(2)Y2d4+2164不对。2165非。2166n=3,l=2,m=0,或±1,或±22167(1)第一个;(2)第一个21682n2=502169(C)2170一个电子的电量(e)改变1伏特(V)电势所需之能量。2171a0或52.9pm2172me或9.109×10-31kg83
2173e或1.602×10-19C2174两个电子相距a0的势能或27.2eV.2175或1.0546×10-34J·s2176(a)n-1,(b)n-3,(c)d轨道有两个径向节面。2177(1)O或核附近(2)a0或52.3pm(3)8×13.6/9eV217802179是2180不是2181不是2182是2183不是21842P3/22185e2186(1)(a)均不能使基态H原子电离,(b)均可使晶体铜中的铜原子电离。(2)=520pm,=416pm。2187在这样的小球体内,概率密度可视为常数当r=10.6pmP=当r=53pmP=2188(1)由于铜晶体中存在着电子与电子,核与核,核与电子之间的相互作用和原子振动,使原子损失能量。其差为(2)该波长在紫外区(3)即高温下从自由原子或晶体发射电子并无差别,此时已是自由电子。2189(C)2190(E)219154.4eV2192对于具有n个电子的体系,其总的自旋量子数S为S=n/2,n/2-1,n/2-2,...,0或1/2设S=n/2-m式中m为非负整数,且m≤n/2多重度=2S+1=n-2m+1因2m恒为偶数,故当n为奇数时,2S+1为偶数。当n为偶数时,2S+1为奇数。21933;0,,;5;2194s态2195相等2196He+中电子,其角动量在x方向上的分量有确定值0,在y方向和z方向上的分量无确定值,平均值为0。2197电子的轨道角动量在x和y方向的分量均无确定值。平均值为0。219810。83
2199==,即-27.2eV.220013.6eV或-13.6eV,2.18×10-18J2201全部;全部;全部2202(C)2203(C)2204(D)2205(B)2206n=3,l=1,│m│=1,又=x故为3px轨道节面数n-1=2个,其中球节面数n-l-1=1个在r=2a0处,角节面数l=1个在yz平面。2207n=3,l=2,│m│=2,=xy故为轨道节面数n-1=2个,其中球节面数n-l-1=0,角节面数l=2个。由=0确定为xz平面,yz平面。2208220962210由Hund规则只能确定能量最低的谱项为5S,他不能排出其它谱项的能级顺序。22112n2;2(2l+1);2;1;22123H,3G,3F,1H,1G,1F。2213(1)15个状态,(2)36个状态22141S,1P,1D,1F,1G.22143S,3P,3D,3F,3G.22152216221722192220222122222223(C)2224(2),(27.211),(2623)2225(B),即电子的静质量。2226(1)态是由和组合而成,,Mz的测量结果中和各占50%,O出现的概率为0。83
(2),在Mz的测量结果中0出现的概率为100%,和出现的概率均为0。(3)出现的概率为100%。0和出现的概率均为0。2227(1)(2)────────1s────(3)∴x=a/Z∴∴(4)────────────1s─────────2228计算公式=E1/(hc)-E2/(hc)主系2p→2s=671.0nm3p→2s=323.4nm4p→2s=274.2nm5p→2s=256.3nm锐系3s→2p=812.9nm4s→2p=497.3nm5s→2p=427.4nm漫系3d→2p=610.5nm4d→2p=460.4nm5d→2p=413.3nm基系4f→3d=1870.2nm5f→3d=1278.5nm2230氢原子光谱第6条谱线产生的光子的能量为83
CH2(CH)6CH2激发所需最低能量为E6>E,能使CH2(CH)6CH2从基态跃迁到第一激发态2231(1)=121nm=92.9nm,Lyman系,紫外光。(2)E1=1.64×10-18Jb>c>d2294(E)2295(A)2296(1)出现的概率,即,Z=2则n=4。出现的概率为(2)概率为(3)83
概率为《结构化学》第三章习题答案3001(A,C)3002Hab=∫a[-Ñ2--+]bdt=EHSab+Sab-∫abdt=EHSab+K因EH=-13.6eV,Sab为正值,故第一项为负值;在分子的核间距条件下,K为负值。所以Hab为负值。3003∫gudt=(4-4S2)-1/2∫(+)((-)dt=(4-4S2)-1/2∫[2-2]dt=(4-4S2)-1/2[1-1]=0故相互正交。3004(C)3006描述分子中单个电子空间运动状态的波函数叫分子轨道。两个近似(1)波恩-奥本海默近似(核质量>>电子质量)(2)单电子近似(定态)3007单个电子3008(B)3009(1)能级高低相近(2)对称性匹配(3)轨道最大重叠3010不正确3011(B)3012=(0.8)1/2fA+(0.2)1/2fB3013能量相近,对称性匹配,最大重叠>,<或<,>3014正确3015不正确3016sppd3017pxpzdxydxzpxp//ppz/s//dxy//d/dxzp//p3018z3019(C)3020p3021s轨道:s-s,s-pz,s-dz,pz–pz,pz-,-,p轨道px–px,px–dxz,py–py,py–dyz,dyz–dyz,dxz–dxzd轨道:dxy-dxy,d-d3022sdp不能不能3023(B)3024原子轨道对分子轨道pz-dxy×83
px-dxzpd-dd-spx–pxp30251s22s21p43s2,3,反磁3026dxy,d3027py,dxy3028C2(1sg)2(1su)2(1pu)2+2s-p混杂显著.因1su为弱反键,而1sg和1pu均为强成键,故键级在2-3之间.3029N2:(1sg)2(1su)2(1pu)4(2sg)2O2:s2s2s2ss2pz2p2px2p2py2p2px*p2py*1或(1sg)2(1su)22sg2(1pu)4(1pg)23030(1sg)2(1su)2(1pu)4(2sg)2的三重键为1个s键(1sg)2,2个p键(1pu)4,键级为3(1su)2和(2sg)2分别具有弱反键和弱成键性质,实际上成为参加成键作用很小的两对孤对电子,可记为:N≡N:。因此N2的键长特别短,键能特别大,是惰性较大的分子。3031O2[KK(sg)2(su*)2(sg2p*)2(pu)4(pg2*)1(pg2*)1]顺磁性C2[KK(sg)2(su*)2(pg2)2(pg2)2]反磁性3032KK(1sg)2(1su)2(1pu)3约3/2[1s22s23s21p44s2]5s22p413033(1)1s22s23s21p41反(2)s1s2s1s2s2s2s2s2s2pz2p2py2p2pz2p2py*2p2px*11.5顺3034p3py,p3pz;p3px3035CN-(1s)2(2s)2(1p)2+2(3s)2键级:33036CFKK-(1s)2(2s)2(3s)2(1p)4(2p)1不论s-p混杂是否明显,最高占据的MO为(2p)1,它是反键轨道。故(C-F)+键强些,短些。3037Cl2:s3s2s3s*s3px2p3py2p3pz2p3py*2p2pz*2反磁性O2+:s2s2s2s*2s2px2p2py2p2pz2p2py*`顺磁性.CN-:-(1s)2(2s)2(1p)4(3s)2反磁性.3038(A),(C)3039(C)3040NF+(1s)2(2s)2(3s)2(1p)4(2p)1键级2.5磁性顺磁性(有一个不成对电子)3041(A)3042OF+>OF>OF-3043EN2>EN2+EO2+>EO2;EOF>EOF-;ECF+>ECF;ECl2+>ECl23044是一个极性较小的分子,偶极矩方向由氧原子指向碳原子。3045OH的HOMO是1s轨道.这是个非键轨道,基本上是O原子的2p轨道.因此,OH的第一电离能与O原子的2p轨道的电离能非常接近。HF的结构与OH类似,它的HOMO是1s轨道,也是个非键轨道,基本上是F的2p83
轨道。因此,HF的第一电离能与F原子2p轨道的电离能非常接近。3046(1)OH基的电子结构为:(1s)2(2s)2(3s)2(1p2py)2(1p2pz)1(2)未成对电子占据p轨道(3)1s轨道是非键轨道,仍保持O原子的2p轨道的特性(4)OH-的最低的电子跃迁的能量比OH基的要高3047H和F以s键结合;HF=N[fH(1)fF(2)+fF(1)fH(2)][aH(1)bF(2)-bF(1)aH(2)]N为归一化系数3048(1)(1s)2(2s)2(3s)2(1p)4(2)HF分子的键长rHF=rH+rF-0.09D,rH,rF是共价半径.=(37+71-0.09×1.9)pm,D=4.0-2.1=1.9=107.8pm3049H2+;;He2+;C2+;NO3050分子N22-O22-F22-N2O2F2电子数161820141618净成键电子数420642F22-净成键电子数为0,不能稳定存在N22-和O22-能稳定存在rN22->rN2rO22->rO23051分子N2+O2+F2+N2O2F2电子数131517141618净成键电子数553642rN22->rN2rO22->rO2rF2+>rF23052分子电子数最高占有分子轨道磁性N214↑↓3sg反O216↑↑2pg顺F218↑↓↑↓2pg反O22+14↑↓3sg反F2+17↑↓↑2sg顺3053COKK(1s)2(2s)2(1p)4(3s)2NOKK(1s)2(2s)2(1p)4(3s2)(2p)1NO在高能反键轨道上有一电子,I1较低。3054[Kr](1s)2(2s)2(3s2)(1p)43055N2+NO;O2+>O2;C2+F2;CN+O2->O2>O2+(3)1865cm-13057比(AB)+的键能小:O2,NO,比(AB)-的键能小:C2,CN83
3058Li2>Li2+;C2>C2+;O2DE丁∴l丁>l乙(3)加热条件,对称性相同,可以环加成。5032(1)分子能级为:E1=a+bE2=aE3=a-b1=1/2f1+/2f2+1/2f32=/2(f1-f3)3=1/2f1-/2f2+1/2f35033(1)E1=a+bE2=aE3=a-b(2)1=0.50f1+0.707f2+0.50f32=0.707f1-0.707f33=0.50f1-0.707f2+0.50f3(3)电荷密度:q1=1q2=1q3=1(4)键序:P12=P23=0.707(5)自由价:C1自由价=1.025C2自由价=0.318C3自由价=1.0255035(A)5037不正确。50360.561.291.4471.724↑↑CH2────CH────CH────CH21.01.083
5038(1).……..…….:N──N──N:有两个互相垂直的∏34.……..…….(2)分子总离域能为1.656b;(3)对每一个∏341=1/2f1+1/f2+1/2f32=1/f1-1/f33=1/2f1-1/f2+1/2f35039ET=6a+2(+1)bDpE=[6a+2(+1)b]-[4(a+b)+2a]=2(-1)b5040(1)Ep=-6.472b(2)ED=2.472b5041E1=a+1.618b;E2=a+0.618b;E3=a-0.618b;E4=a-1.618bDE=E2-E1=-b=11.46-9.03=2.43(eV)b=-2.43eV5042.H==C==H中两个小p键彼此垂直,即:z方向上--C==C,存在∏33;y方向上C==C--,也存在∏33。z方向:P12=P23=2××/2=/2,同理y方向得P12’=P23’=/2再考虑两个s键键级得:2+4(/2)=4.828>4.7325048(A),亲核反应发生在电荷密度最小处1位。5049DpE=1.64b5050ri=1.00P12=P23=P34=P45=P56=P61=0.667Fi=0.3995051HOMO:3=1/(f1-f3+f5);节面通过C2,C4,垂直键轴,共2个。LUMO:4=(f1-f2+f4-f5);节面有3个,均垂直键轴,通过C1--C2中点,C3和C4--C5中点。50523节面通过C3和C1-C5中点。5053该共轭分子的s骨架为:(略)5054离域能为050551─2─3─4─5=1/2(f1+f2-f4-f5)5056∏34;Cs。5057(1)2个∏44(2)2个∏56(3)∏78(4)∏910(5)∏465058(E)5059(A)5060NO2NO2+NO2-∏34(或∏33)2∏34∏34键强次序NO2+>NO2>NO2-5061C2H2Cl2有三种异构体:HClHHHCly///↑(1)C==C(2)C==C(3)C==C─→x///ClHClClHCl(1)m=0;(2)m=(mC-Cl+mC-H)(在y方向)(3)m=mC-Cl+mC-H(在x方向)mC-Cl为C-Cl键键距,mC-H为C-H键键距,偶极距大小顺序为(2)>(3)>(1)5062对。5063乙烯HOMO与Br2LUMO(s4pz*)对称性不匹配;乙烯LUMO与Br2HOMO(p4p*)虽对称性匹配,但电负性xBr>xCBr2的反键轨道提供电子,加强Br--Br键,不易断裂。5064是。5065(1)放热,3s+p→4s;(2)放热,4s+2p→6s。5066顺。5067因无催化剂时对称性不匹配;用Ni作催化剂,将吸附H2变成H原子成为占有电子的轨道,和乙烯的LUMO对称性匹配。5068是。5069(C)83
5070设它们的力常数k基本相等,De都一致,则D0为:De/eVmh0/eVD0/eVH24.721/20.264.46D24.7210.184.54HD4.722/30.234.49T24.721.50.154.575071242kJ/mol1250kJ/molCl2─→Cl+Cl─→Cl+Cl++eˉ↓1085E↑Cl2+──────────`若E为Cl2+的键能,按上式循环可得:E=(242+1250–1085)kJ/mol=407kJ/mol5072(B)1=p-1/2cosnf,2=p-1/2sinnf5073En=;r=140pm5074(1)平面正方形;(2)正四面体;(3)三角锥;(4)T形;(5)四方锥。5075SO32-:三角锥形,C3v;SO3:平面三角形,D3h;XeOF4:四方锥形,C4v;NO2+:直线形,D∞h;NO2:弯曲形,C2v5076[20×154×cos(109.5°/2)+400]pm=2916pm5078(C)5079(B)5080H==2.35T5081(D)5082(A)5083CH2本身两个氢核间有很强的自旋偶合作用,它会引起核磁能级位移,但谱线的频率保持不变,所以谱峰不会进一步分裂。由于H+的存在,OH发生快速交换,使OH质子对CH2的影响平均化,OH质子也不会引起CH2质子谱峰进一步分裂。50842I+1,1H和aC5085(D)5086(B)5087(C)5088(B)5089CH4:C是sp3杂化,键角109.5°;NH3:N也是sp3杂化,因有一对孤对电子,对成键电子对有排斥;H2O:O是sp3杂化,但有两对孤对电子,对成键电子对排斥更大;5090NO2-角形(键角<120°=;NO2+直线形;Xe平面四方形。(CH3)2SnSn原子处于由2个C原子和2个F原子构成的四面体中心,每个C原子处于由3个H原子和Sn原子构成的四面体中心。CO(NH2)2三角形,但双键占有较大空间。∠OCN>120°∠NCN<120°5091(A)5092(A)5093电负性:N>P>As>Sb使XH与XH间的排斥作用依次减小。5094见周公度《结构化学基础》p.244。5095羧酸、苯酚电离出H+后形成离域p键,故能稳定存在。∏34∏78∏78∏345096丁二烯,∏44;BF3,∏46;[CH2=CH─CH2]+,∏325097sp杂化;2个∏34。5098T型比较稳定。5099见周公度《结构化学基础》p.204。83
5100C2的HOMO(1pu与H2的LUMO(s1s*)对称性不匹配。H2的HOMO(s1s)与C2的LUMO(2sg)对称性不匹配,此反应不能进行。5102一个氨基酸的氨基与另一个氨基酸的羧基缩合,失去一分子水而生成的酰胺键称为肽键。5103C--O键长次序:CH3COCH3>CO2>CO,理由从略。5104(a)∏1518(b)∏88(c)∏88(d)∏46(e)∏10125105N--C—C--N,C:sp杂化,与N的一个p轨道生成s键,与另外一个C原子的sp杂化轨道生成s键,C和N剩下的两个p轨道可以形成两个∏44;四个p轨道组成四个分子轨道,其中两个是成键轨道,两个是反键轨道,能量最低的成键轨道没有节面,中心反对称,另一个成键轨道有一个节面,中心对称;基态时4个p电子填充在两个成键轨道上,电子全部配对,分子是反磁的。5106(B)5107石墨层内形成∏nn。而(BN)x虽满足形成离域p键的条件,但由于B--N的极化作用,使其能带分成两个亚带,禁区宽度很大(440kJ·mol-1),无离域能出现。5108根据4n+2规则,从七元环移走一个电子到五元环,使两者皆有6个p电子,稳定,故七元环显正电性,五元环显负电性。5109(a)净重叠为0(b)前线轨道对称性匹配,较稳定。5110苯∏66氯乙烯∏34丙烯基阳离子∏325111NH3:N采用不等性sp3杂化,呈三角锥形。BCl3:B采用sp2杂化,有一空的p轨道,可以接受NH3的孤对电子形成分子配合物,生成配合物时B采用sp3杂化,配合物呈双三角锥型。5112(a)八面体sp3d2(b)平面四方形dsp2(c)平面三角形(d)平面三角形sp2(e)三角锥不等性sp35113(a)∏88(b)∏910(c)∏995114(a)∏88(b)∏910(c)∏87(d)∏895115(a)∏910(b)∏1012(c)∏9105116(a)∏33(b)∏78(c)∏88(d)∏1414(e)∏465117三个化合物均有∏66,失去一个氯原子后,p键扩大为∏76,∏1213,∏1918,体系能量降低,所以,7失去氯原子较有利,因此Cl显得活泼。由于大p键能∏1918>∏1312>∏76,所以,(c)中的氯最活泼。5118(e)>(c)>(b)>(d)>(a)5119N采用sp2不等性杂化,与两个O形成两个s键,余下的p电子组成∏33,有未成对电子,具有顺磁性。5120BF3:B采用sp2杂化,与三个F形成三个s键。B的一条空轨道与三个F的p轨道形成∏46。平面三角型。NF3:N采用sp3不等性杂化,与三个F形成三个s键。三角锥型。5121(B)是稳定构型5122∏y34∏z34图略51230r=a0≤f≤2p(1)=-+V,=E,V=∞r≠a0≤f≤2pEn=,0=,n=cosnf,n=sinnfn=0,±1,±2,···(2)···──────n=±2────────n=±1──────n=0──环势箱模型HMO法(3)如果大p体系有4m+2个电子,由能级图知:满壳层,稳定。(4)lmax=2094A5124在R4NOH分子中的N上已经没有可供与H2O中的O形成氢键的氢存在,不可能形成N--H···O氢键,取而代之的是斥电子基R,故N上给电子能力很强,即碱性很强,甚至比NaOH还要强。R3NHOH中,N原子上有一可与O形成氢键的H,N原子上孤对电子给电子能力减弱,即碱性减弱。在83
NH4OH分子中,N原子连接的可形成氢键的H较多,而N上的孤电子对必须用来供给其N-H基生成的氢键N--H···O,其给电子能力很弱,即为弱碱。5125(A)5126(B)吸收最短波长的光,(C)吸收最长波长的光。5127H3CCl中C为sp3杂化,C--Cl为单键。H2C==CHCl中C为sp2杂化,C--Cl间除单键外还有大p键∏34,所以其中C--Cl键键长较H3CCl中C--Cl键键长短。HC≡CCl中C--Cl间除s键外,还有两个p键∏34,所以其C--Cl键最短。5128由多于两个原子轨道线性组合而成的分子轨道称为离域分子轨道。处于离域分子轨道上的电子不再局限在某两个原子间运动,而是更平均地围绕分子中更多的原子运动。5129S8有八个S--S键,故S--S键能为ES-S=2130/8kJ·mol-1=266kJ·mol-1H2S中有两个H--S键,故H--S键能为EH-S=735/2=368kJ·mol-1H2S2中有两个H--S键,一个S--S键,故上述反应所需能量为:E=(266+368×2)kJ·mol-1=1002kJ·mol-1.与实验值接近,多1.6%.5130把由HMO法求得的共轭分子的电荷密度ri,键级Pij,及自由价Fi标在其分子结构图上,并且规定ri写在第i个C原子旁,Pij写在第i,j个C原子间的化学键上,Fi写在由第i个原子引出的箭头上.所得如下的图形叫分子图。FiFJ↑Pij↑──Ci───CJ──rirJ5131H2+O2→H2O2反应前后键的变化是Hf=EH-H+EO=O-2EH-O-EO-OEO-O=EH-H+EO=O-2EH-O-DHfO2分子为双键,故不能作为O--O单键的解离能。5132前者存在空间阻碍,使得两个苯环不在一个平面内,破坏了两个苯环的共轭,形成两个∏66,因而其光谱和苯相似。后者无空间阻碍,两个苯环在同一个平面内,形成∏1212,p电子活动范围增大,故紫外光谱吸收波长比苯长。5133(a)中大p键为∏66,(b)中大p键为∏1010。当电子活动范围增大时,按一维势箱能级公式E=n2h2/8ma2`和平面刚性转子能级公式E=n2h2/8mp2r2,相邻能级差变小,紫外可见光谱向低频方向移动,所以(a)比(b)波数大。5134以上三个化合物的紫外可见光谱都是由于p电子跃迁产生的。苯具有大p键∏66,而苯胺中的大p键为∏78,两者p电子能级不同,能级间隔不同,因而紫外可见光谱不同。苯胺的盐酸盐C6H5NH3Cl中,N上电子不再参与大p键,故其大p键与苯相同,为∏66,从而其光谱也相近。5135CH3--CH2CH2CH2CH2--CH3→+H2反应前后键的变化是5C--C+14C--H→6C--C+12C--H+H--H,可简化为2C--H→C--C+H--HDH=2EC-H-EC-C-EH-H=(2×415-344-436)kJ·mol-1=50kJ·mol-1DH>0,为吸热反应。5136CH3--CH2--CH2--CH2--CH2--CH3→CH3--CH(CH3)--CH2--CH2--CH3因前后无键的变化,所以DH=05137C6H10+H2→C6H12反应前后键的变化为C==C+5C--C+10C--H→6C--C+12C--H可简化为C==C+H--H→C--C+2C--HDH=EC=C+EH-H-EC-C-2EC-H=(615+436-344-2×415)kJ·mol-1=-123kJ·mol-1DH<0,为放热,放热123kJ·mol-1。51383HC≡CH→C6H6将苯近似看作单双键相隔,反应前后键的变化为3C≡C+6C--H→3C--C+3C==C+6C--H可简化为3C≡C→3C--C+3C-=CDH=3EC≡C-3EC-C-3EC=C=[3×(812-344-615)]kJ·mol-1=-441kJ·mol-1DH<0,为放热反应。5139C6H12→C6H6+3H2将苯近似看作单双键相隔,反应前后键的变化为6C--C+12C--H→3C--C+3C==C+6C--H+3H--H可简化为3C--C+6C--H→3C==C+3H--HDH=3EC-C+6EC-H-3EC=C-3EH-H=(3×344+6×415-3×615-3×436)kJ·mol-1=369kJ·mol-183
DH>0,为吸热反应,吸热369kJ·mol-1。5140丙酮中C和O之间为双键,CO2中为共轭大p键。CO中为一个s键,一个p键,一个配键,成键电子数为6,相当于三键。5141(a)∏88(b)∏77(c)∏910(d)∏34(e)∏345142(a)∏1414(b)∏88(c)∏910(d)∏1620(e)∏345143(a)无(b)∏34(c)∏88(d)∏1414(e)∏465144(a)∏x44∏y44(b)∏88(c)∏90(d)∏1012(e)∏465145Cl的活泼性:(d)>(c)>(b)>(a)5146成键情况和理由略。碱性:(b)>(a)>(c)>(d)51471.三种异构体;2.(1)中Ha之间,Ha’之间通过C2相互变换;Ha和Ha’通过镜面相互变换,因此四个质子是等价的。只有一个1HNMR讯号。(2)HC和HC’是等价的,三个1HNMR讯号。(3)两个Ha,两个Hb通过C2相互变换,从而两个Ha等价,两个Hb等价。但Ha和Hb没有对称操作可以相互变换,它们不等价。有两个1HNMR讯号。5148两组等性质子(1,2),(3,4,5,6)ESR谱线数目=(2×2×+1)×(2×4×+1)=15条由理论杆谱得出的相对强度比为1:2:1:4:8:4:6:12:6:4:8:4:1:2:151491.两种异构体;2.两种异构体(1)和(2)都分别只有一类质子,因此只有一个1HNMR讯号。5150室温时一个未成对电子可以在两个铜核之间跃迁,电子受两个铜核的作用,ESR讯号分裂为2×2×+1=7条谱线。在低温时没有足够的热能使大环的构型转变为对电子在两个铜核之间跃迁有利的构型,因此,电子定域在一个铜核上,受一个铜核的影响,ESR谱线分裂为2×1×+1=4条。5151(B)(E)5152(D)5153强度比为1:2:2:15154特种大p键的生成(超共轭效应)使p电子体系的离域范围扩大,能级间隔变小,紫外吸收频率变小。甲苯比苯红移8nm,二甲苯比苯红移16nm。5155苯分子的离域能等于乙烯p键电子能量的三倍与苯分子的离域p键电子能量之差,即3×2(a+b)-[4(a+b)+2(a+2b)]=-2b5156写出六个久期方程;利用对称性将系数关系列出;按四种方式(SXSy,SXAy,AXSy,AXAy)简化久期方程并解之,求出六个x值;将各x值代入E=a-xb,求出六个E值;按定义和电子排布情况求出离域能1.656b。5157如果顺磁共振只有未成对电子与外磁场作用,那么顺磁体系的ESR谱将只有一条谱线,其区别在于g因子不同而已。实际上顺磁体系中还有磁性核,在磁场中电子自旋与核自旋相互作用,引起谱线分裂,这称为超精细结构。在HO2中一个未成对电子,ms=±1/2在磁场中分裂为两个能级而产生一个共振峰,还有一个质子I=1/2,mI=±1/2.质子核磁矩在磁场中取向不同相当于施了两个方向的附加磁场,如磁矩顺着磁场方向,即增大了电子所在处的磁场强度,使外磁场略低于原来的磁场,而产生共振峰如磁矩逆着外磁场方向,则需要略高于原磁场才能发生共振吸收,所以,一条谱线分裂为两条谱线。5158形成离域p键的体系的能量比相应的经典结构式所表达的普通单双键体系应有的能量低,这降低的能量称为离域能。5159CH3F:a=0.2638(∠H--C--H)CH2F2:a=0.2716(∠H--C--H)CH2F2:a=0.2391(∠F--C--F)CHF3:a=0.2437(∠F--C--F)5160ge=1.985161化学位移d,ppm,质子所属的基团和种类;偶合常数J,Hz,各类质子的相对位置;峰面积,各类质子数目之比。5162(略)5163直线型:a,b,g三角双锥:c平面三角形:d83
平面型:e,f弯曲型:h,iN───N:j//5164==exp[(Eb-Ea)/kT](1)核磁共振条件:E=Eb-Ea=hl(2)将(2)式代入(1)式得:=exp[h/kT]=1.00001由于a态上的质子仅比b态上的质子多出十万分之一,而跃迁概率则与自旋状态的质子数成正比,因此实验测量的是一个非常微弱的净的吸收讯号。51650=rH0/2p1=rH1/2p=rH0(1-s1)/2p=0(1-s1)2=rH2/2p=rH0(1-s2)/2p=0(1-s2)D=1-2=0(s2-s1)d1=(sR-s1)×106,d2=(sR-s2)×106Dd=d1-d2=(s2-s1)×106∴D=0×10-6×Dd当0=60MHz时,D=60Hz当0=100MHz时,D=100Hz5166(D)5167正确5168(1)l=42.57MHz(2)l=60.00MHz5169六个峰。5170所有的芳环质子处于负屏蔽区,(a)和(b)应一样。但(a)位质子羰基的诱导效应,使它的屏蔽作用更小,所以t值较小,d值较大。5171略5172(C)5173(A)5174(A)5175(A)5176/x10/c1½1x1½½c2½=0r1=r3=2×(1/2)2+1×(1/)2=1r2=2×(1/)2=1�1x/c3/P12=P23=2×(1/2)×1/=0.7075177由∫12dt=0得1/3+c2/=0c2=-1/再由∫32dt=1,1/3+1/6+c32=1c32=1-1/3-1/6=1/2c3=±1/∫23dt=0取c3=-1/∴c2=-1/c3=-1/5178将从基态原子形成某一分子时的生成热焓(温度为298K)看作是该分子中所有各键的键能之和来定义键能,并根据实验或计算所得的热焓来推算键能。同一化学键的键能与其化学环境有关。手册中列的键能是参照各种化合物中同类键的键能平均值。键能是化学键强度的一种量度。5179O2→2OH1=498kJ/mol①O→O++e-H2=1311kJ/mol②O2→O2++e-H3=1165kJ/mol③DH=DH1+DH2-DH3=644kJ/molO2+→O+O+5180N2→2NDH1=945kJ/mol①N→N++e-DH2=1400kJ/mol②N2→N2++e-DH3=1503kJ/mol③DH=DH1+DH2-DH3=842kJ/molN2+→N+N+5181F2→2FDH1=158.3kJ/mol①F→F++e-DH2=1629kJ/mol②F2→F2++e-DH3=1515kJ/mol③①+②-③F2+→F+F+DH=DH1+DH2-DH3=322.3kJ/mol5182夹角为109°28"5183分子中某一个键的解离能是指仅仅断裂这个键,即分子分裂为原来这个键所连接的两个部分时所需要的能量。5184通常规定A--A共价键键长的一半为原子A的共价半径,又称原子半径。它不是单个原子的半径,83
而是原子在成键时所显示出来的大小,因为单个原子周围的电子云无确定的边界。5186分子间以范德华力相结合时,不同分子的相互接触的原子表现出的半径,称为范德华半径,它是按照相互接触的原子间的距离即为该两原子的范德华半径之和而推算出来的,范德华半径比共价半径大,变化范围也大,即守恒性差。5187(B)5188分子SO3CO2Co(en)3点群D3hD∞hD3杂化轨道sp2spd2sp3离域p键∏462∏34无偶极矩无无无旋光性无无有51892CC3x-100-10c1½½-1x0000c21CC400x-100c3=0/00-1x-10c4C5-100-1x-1c5½0000-1xc6C6利用归一化条件,c12+c22+c32+c42+c52+c62=1得E2能级分子轨道2=0.5(f1+f2-f3-f4)5190x=1.9319,E1=a+1.9319b为最低能级,带入方程(按下图编号)2CC3½½1CC4/C5½C6/x-100-10/c1½-1x0000½½c2½½00x-100½½c3½=0利用归一化条件,c12+c22+c32+c42+c52+c62=1½00-1x-10½½c4½得最低能级分子轨道½-100-1x-1½½c5½1=0.444f1+0.230f2+0.230f3+0.444f4+0.628f5+0.325f6�000-1x/c6/5191按下图编号2CC3½½1CC4/C5½C6x-100-10-1x0000x-100-1x-100083
00x-100-1x000-1x00000-1x-10=x00x-10+00x-10-100-1x-100-1x-100-1x00000-1x-100-1x-100-1-1x-10-1x-10-1x-100=x-1x00+x-1x00--1x0000x-100-1-100x-1-10-1x-100x00-1xx-1-1x-10x-1-1=x3-1x0+x2-1x0-x-1x0-(x2-1)2-10x-10-1-10x=(x3-x)(x3-x-x)-x2(x2-1)-(x2-1)2=(x2-1)(x4-4x2+1)=0x=±1,x=±0.5176,x=±1.9369E1=a+1.9369b,E2=a+b,E3=a+0.5176b,E4=a-0.5176b,E5=a-b,E6=a-1.9319b5192因b<0,共6个p电子,最高占有轨道为E3=a+0.518b,即x=0.518将x=0.518带入方程(按下图编号)2CC3½½1CC4/C5½C6/x-100-10/c1-1x0000c200x-100c3=000-1x-10c4-100-1x-1c5\0000-1x/\c6/利用归一化条件,c12+c22+c32+c42+c52+c62=1,得3=0.230f1+0.444f2+0.444f3+0.230f4-0.325f5-0.628f65193x=2,E2=a+xb=a+2b为最低能级,将x=2代入方程(按下图编号)/x-1000–1/c1-1x-1000c20-1x-100c3=000-1x-10c4000-1x–1c5-1000-1x/c6/利用归一化条件,c12+c22+c32+c42+c52+c62=1得=(f1+f2+f3+f4+f5+f6)5195(1)三种可能的异构体(2)(a)和(b)有三组等价质子,产生三个1HNMR讯号。(c)只有一个对称面,有6类不同的质子,产生683
个1HNMR讯号。5196四甲基硅5197(A)5194该体系有4个p电子,基态只占据最低能量的两个分子轨道。E1=a+b,将x=带入方程(按下图编号)·──·──·──·──·12345x-1000c1-1x-100c20-1x-10c3=0①00-1x-1c4000-1xc5利用归一化条件,c12+c22+c32+c42+c52=1得1=0.2887f1+0.5f2+0.5773f3+0.5f4+0.2887f5将x=1代入方程组①,且∑c12=1,得2=0.5f1+0.5f2-0.5f4-0.5f5,分子图为:0.5770.3660.9430.78870.5773↑↑↑·────·────·────·────·0.671.00.675198S采用sp3d2杂化,与6个F形成共价键,分子对称性较高,无极性,故有良好的绝缘性,未成对电子数为0,是反磁性化合物。5199∏1616∏10145200分子SO42-O3SO3SF6中心原子的等性sp3不等性sp2等性sp2sp3d2杂化方式几何构型正四面体V型正三角型正八面体点群TdC2vD3hOh5201(C)5202(60×Dd+2.5J)×2p/gN=34.4×10-3高斯①(100×Dd+2.5J)×2p/gN=54.5×10-3高斯②②-①(100-60)Dd×2p/gN=20.1×10-3Dd=20.1×10-3×26753/(2p×40)=2.14ppmJ=[34.4×10-3gN/2p-60×Dd]/2.5=(146.47-128.4)/2.5=7.24ppm=7.24×2p/gN=1.7×10-3高斯=7.2285202(60×Dd+2.5J)×2p/gN=34.4×10-3高斯①(100×Dd+2.5J)×2p/gN=54.5×10-3高斯②②-①(100-60)Dd×2p/gN=20.1×10-3Dd=20.1×10-3×26753/(2p×40)=2.14ppmJ=[34.4×10-3gN/2p-60×Dd]/2.5=(146.47-128.4)/2.5=7.24ppm=7.24×2p/gN=1.7×10-3高斯=7.2285204略5205(A)5206(1)2nI+1=2×4×(1/2)+1=5条(2)2nI+1=2×6×(1/2)+1=7条(3)(2n1I+1)(2n2I+1)=(2×4×+1)(2×4×+1)=25条5207加成产物为a。其HNMR谱中两个三重峰分别归属于两个--CH2--,两个单峰分别归属于两个--CH3。5208烯醇式的百分含量=37/(37+19.5)×100%=65.5%d=3.66峰属于--CH2--,d=5.62峰属于--OH(或—CH==)5209正确。由环辛四烯负离子基的ESR谱可知其八个质子是等性的。即ESR谱线数目为(2×8×+1)=9条,其强度比为:C08:C18:C28:C38:C48:C58:C68:C78:C88=1:8:28:56:70:56:28:8:1因此,环辛四烯负离子基是平面构型。5210酚酞在碱性溶液中发生下列反应,在反应过程中碳原子C*发生轨道改组(sp3→sp2),使产物分子中p电子的离域范围扩大,能级间隔变小,吸收频率移到可见光区。83
5211配合物Cu(Ⅱ)Cu(Ⅰ)L,有一个未成对电子,这个未成对电子可以在两个Cu核之间跃迁,ICu`=2/3.受磁性核的影响,ESR讯号产生超精细分裂,谱峰数目等于2nI+1=2×2×(2/3)+1=7。暴露给CO后,Cu(Ⅰ)易与CO生成加合物,加合物的生成影响了络合物的构型变化,从而影响了电子在两个Cu核之间的跃迁。未成对电子定域在一个Cu核上,谱峰数目等于2×(2/3)+1=4.当电子在两个Cu核之间跃迁时,每个Cu核上的电子密度约为0.5,因此与Cu核的超精细相互作用较弱,A为40--45G,约为定域在一个Cu核上的一半,后者A为85G。5212中心氧原子采取不等性sp2杂化,形成三个杂化轨道,其中两个杂化轨道与端基氧原子形成键,孤对电子占据第三个杂化轨道;中心O原子还有一个未杂化的P轨道与两端的氧原子的P轨道形成,可见中心O原子的电子贡献多,两端的氧原子得部分负电荷。因此,O—O键为极性键,同时分子构型是V型,正负电荷重心不重合。所以,O3是极性分子。5213苯吸收低能量的光,因苯中电子的跃迁所需的能量小于乙烯中电子的跃迁所需的能量。5214乙烷分子,因其电离的是电子,乙烯分子电离的是电子。5215等性sp3杂化,则有(1)杂化轨道的归一性,则有。5216(A)5217只有1个峰,表明所有碳原子有完全相同的周围环境,这些碳原子只能排布在一个球面上。另外,六方晶胞中c/a=1.63,进一步说明C60分子呈圆球形。5218CH3COCl形成分子间氢键C—H····O。52195219(E)83'
您可能关注的文档
- 《细胞生物学》习题及解答.doc
- 《经济博弈论》课后答案、补充习题答案.doc
- 《经济地理学 第二版》课后思考题答案.doc
- 《经济地理学》第二版课后思考题答案.doc
- 《经济地理学》课后思考题答案.doc
- 《经济学基础》习题试卷与答案.doc
- 《经济学基础》各讲习题及参考答案(简).doc
- 《经济应用数学—概率论与数理统计》马统一的习题1一5答案.doc
- 《结构力学》课后习题答案文治国版 重庆大学出版社.pdf
- 学基础习题》答案_周公度_第4版.doc
- 《绝密文档!!!》电梯练习题(附答案).doc
- 《统计学(专业版)》2012-习题集-解答-陈正伟.doc
- 《统计学》(第四版)学习指导书以及课后习题答案.doc
- 《统计学》(贾俊平,第五版)分章习题及答案.doc
- 《统计学》2011-12-习题集-解答-定稿-打印.doc
- 《统计学》习题集及答案.doc
- 《统计学》课后习题答案.doc
- 《统计学》课程的教学教案.doc
相关文档
- 施工规范CECS140-2002给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程
- 施工规范CECS141-2002给水排水工程埋地钢管管道结构设计规程
- 施工规范CECS142-2002给水排水工程埋地铸铁管管道结构设计规程
- 施工规范CECS143-2002给水排水工程埋地预制混凝土圆形管管道结构设计规程
- 施工规范CECS145-2002给水排水工程埋地矩形管管道结构设计规程
- 施工规范CECS190-2005给水排水工程埋地玻璃纤维增强塑料夹砂管管道结构设计规程
- cecs 140:2002 给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程(含条文说明)
- cecs 141:2002 给水排水工程埋地钢管管道结构设计规程 条文说明
- cecs 140:2002 给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程 条文说明
- cecs 142:2002 给水排水工程埋地铸铁管管道结构设计规程 条文说明