

- 411.48 KB
- 2022-04-22 13:43:31 发布


- 1、本文档共5页,可阅读全部内容。
- 2、本文档内容版权归属内容提供方,所产生的收益全部归内容提供方所有。如果您对本文有版权争议,可选择认领,认领后既往收益都归您。
- 3、本文档由用户上传,本站不保证质量和数量令人满意,可能有诸多瑕疵,付费之前,请仔细先通过免费阅读内容等途径辨别内容交易风险。如存在严重挂羊头卖狗肉之情形,可联系本站下载客服投诉处理。
- 文档侵权举报电话:19940600175。
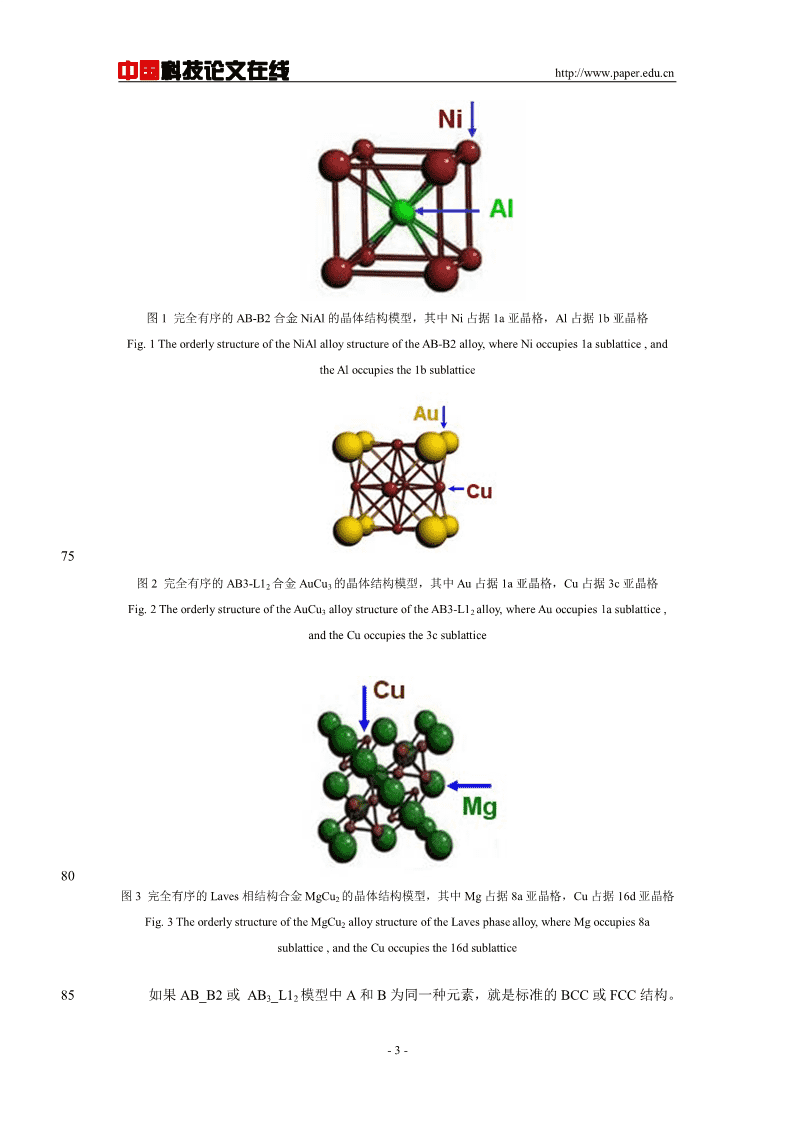
'中国科技论文在线http://www.paper.edu.cn多主元高熵合金MoNbTaVW中合金元素的占#位行为**谢哲宇,吴波,黄锦长,杨义许,李皎亮5(福州大学材料科学与工程学院,福州350100)摘要:本文基于严格的晶体学结构信息,运用合金热力学计算模拟,理论预测了多主元高熵合金MoNbTaVW的元素占位行为随温度的变化规律。基于占位分数,建立原子在亚晶格上的分布构型,进而利用第一性原理方法预测合金相的热力学和力学性质。MoNbTaVW合金在873K到2373K温度范围内为BCC相。不同的合金元素占位倾向性不相同,其中Mo原10子基本占据1a亚晶格位置,Nb原子基本占据1b亚晶格位置;而W、Ta和V原子在两种亚晶格上的占位分数随温度的变化较大,随温度的升高,W原子逐渐由1a亚晶格向1b亚晶格转移,Ta原子和V原子由1b亚晶格位置,向1a亚晶格位置转移。尽管MoNbTaVW合金的元素占位情况随温度发生变化,但平衡体积随温度变化不大,体积弹性模量也相差不大,约为223GPa,因此,MoNbTaVW合金具有高温稳定性特征。15关键词:MoNbTaVW;第一性原理计算;合金热力学;占位行为;力学性质中图分类号:TG111.5PredictionofthesiteoccupyingbehaviorofMulti-principalhigh-entropyalloyMoNbTaVW20XIEZheyu,WUBo,HUANGJinchang,YANGYixu,LIJiaoliang(SchoolofMaterialsScienceandEngineering,FuzhouUniversity,Fuzhou350100)Abstract:Basedonthestrictcrystallographicstructureinformation,usingalloythermodynamicssimulation,thetemperature-dependenceofsiteoccupyingfractionsofMoNbTaVWhighentropyalloywereobtained.Basedonthesiteoccupyingfractionsofelementsonthesublattices,theatom25occupyingconfigurationwereestablishedandthethermodynamicandmechanicalpropertiesofalloywerepredictedusingfirst-principlescalculationsbasedondensityfunctionaltheory(DFT).MoNbTaVWalloyshowBCCphaseinthetemperaturerangefrom873Kto2223K.Thesitepreferencesofdifferentatomsarequitedifferent.Moatomsbasicallyoccupysublattice1a;Nbatomsbasicallyoccupysublattice1b,whilethesitepreferencesofW,TaandVatomsaredependentonthe30heattreatmenttemperature.Withtheincreasesoftemperature,Watomsgraduallytransferfromsublattice1atosublattice1b,whileTaandVatomsgraduallytransferfromsublattice1btosublattice1a.Althoughthesiteoccupyingbehaviourschangewiththeheattreatmenttemperature,theequilibriumvolumeandbulkelasticmoduluschangelittleatdifferenttemperature,whichrevealthatMoNbTaVWhighentropyalloyhasgoodtemperaturestability,andthebulkelasticmodulusisabout35223GPa.Keywords:MoNbTaVW;first-principlescalculations,thermodynamics,siteoccupyingfraction,mechanicalproperties400引言传统合金的设计原则一般以一种或两种合金元素为主元,其成分超过50at.%。合金化改[1,2]性元素添加量不超过35at.%,以获得良好的组织和性能。高熵合金又称为多元高混乱度基金项目:国家自然科学基金(No.50971043,51171046);教育部博士点基金(博导类)(No.20133514110006)作者简介:谢哲宇(1993-),男,硕士研究生,材料计算与模拟通信联系人:吴波(1971-),男,教授,博士生导师,材料计算与模拟.E-mail:wubo@fzu.edu.cn-1-
中国科技论文在线http://www.paper.edu.cn[3-11]合金,就是主要元素(简称主元)是在5~13种之间,每种元素原子百分比5%<35%,也可称之为无主元合金。多主元高熵合金设计理念突破了传统合金在成分设计方面的禁锢,45尝试四主元以上的多主元,甚至等摩尔比进行合金设计,一般认为这样的成分特征使得合金的混合熵很高,可望进一步使得相结构稳定和简单,并带来一种或多种特殊物理、力学和化[12]学性能。MoNbTaVW合金是一个具有良好的硬度、强度和优秀耐磨性的高温材料。通过对比MoTaWNbV与商业合金(铬镍铁合金718)的磨损行为,表明其作为耐磨性高熵合金具有诱人的前景,并且预期其高温性能亦非常可观。因此,高熔点高熵合金的设计和制备具[12-15]50有重要理论意义和应用价值。通常认为多主元高熵合金原子在点阵上的排列接近理想混合,用玻尔兹曼公式计算混合熵,但是,合金中元素在亚晶格上的占位存在或多或少的有序化(倾向性),本文拟从原子占位的角度,探索BCC高熵合金MoNbTaVW形成的热力学原理,计算出一定多主元体系的熵实际能“高”到什么程度,探讨稳定构型,并用于合金性能的第一性原理计算模拟。551计算方法1.1热力学亚晶格模型由于高熵合金多数是呈现FCC和BCC结构,同时可能析出少量的C15结构的Laves[16]相,L10、D03和D019等结构。若将所有结构均加以考虑,计算量非常大,相平衡求解也是相当困难的。作为合金设计方法的前期探索,本论文拟先对下列三种结构优先考虑,即以60NiAl为原型的AB_B2结构(注:当A和B为同种元素,或合金为完全无序的固溶体,则结构就等效为BCC结构),以AuCu3为原型的AB3_L12结构,以及以MgCu2为原型的AB2_C15结构(即Laves相)。后续研究工作将会进一步扩大相的种类,最终将多主元体系中,所有可能存在的合金相结构都考虑进去,逐一建立热力学亚晶格模型,最后建立起一个多元多相合金热力学数据库。65以NiAl为原型的AB_B2结构,以AuCu3为原型的AB3_L12结构,,以及以MgCu2为原型的AB2_C15结构(Laves相)的晶胞结构如图1至图3所示,其中AuCu3为原型的AB3_L12结构中Au占据顶角位置(1a亚晶格),Cu占据面心位置(3c亚晶格);以NiAl为原型的AB_B2结构中Ni占据顶角位置(1a亚晶格),Al占据体心位置(1b亚晶格);以MgCu2为原型的AB2_C15结构中Mg占据8a亚晶格,Cu占据16d亚晶格。70-2-
中国科技论文在线http://www.paper.edu.cn图1完全有序的AB-B2合金NiAl的晶体结构模型,其中Ni占据1a亚晶格,Al占据1b亚晶格Fig.1TheorderlystructureoftheNiAlalloystructureoftheAB-B2alloy,whereNioccupies1asublattice,andtheAloccupiesthe1bsublattice75图2完全有序的AB3-L12合金AuCu3的晶体结构模型,其中Au占据1a亚晶格,Cu占据3c亚晶格Fig.2TheorderlystructureoftheAuCu3alloystructureoftheAB3-L12alloy,whereAuoccupies1asublattice,andtheCuoccupiesthe3csublattice80图3完全有序的Laves相结构合金MgCu2的晶体结构模型,其中Mg占据8a亚晶格,Cu占据16d亚晶格Fig.3TheorderlystructureoftheMgCu2alloystructureoftheLavesphasealloy,whereMgoccupies8asublattice,andtheCuoccupiesthe16dsublattice85如果AB_B2或AB3_L12模型中A和B为同一种元素,就是标准的BCC或FCC结构。-3-
中国科技论文在线http://www.paper.edu.cn当每种亚晶格分别为同一种或不同种元素完全占据,这样形成的占位完全有序的化合物称为端基化合物(End-memberCompound)。对于多元合金,根据晶体学结构信息,采用相应的亚晶格模型描述,表达式(1)、(2)和(3)分别给出了AB_B2型BCC结构、AB3_L12型FCC结构和AB2_C15型Laves相结构的双亚晶格模型:90(EE12yy11aa,,...,EEEiiyyy1a)(1211bb,,...,Ey1b)(1)EE12EiiEE12E(EE12yy11aa,,...,Eiiy1a)(EE12yy33cc,,...,Ey3c)(2)EE12Eii13EE12E(EE12yy88aa,,,Eiiy8a)(EE12yy161dd,,6,Ey16d)(3)EE12Eii12EE12E95其中,1a、1b、3c、8a和16d为亚晶格名称,即a、b、c和d为Wyckoff位置,1、3、1a1b3c8a16d8和16为多重性因子。yEi、yEi、yEi、yEi和yEi为Ei元素分别在1a、1b、3c、8a和16d亚晶格上的占位分数。1.2热力学模型求解过程AB_B2型BCC结构的Gibbs自由能的求解转化为一个两步走的计算途径,如图4所示。100①MoNbTaVW,,,,(,,,,)→Mo111aaaNbTaV1aW1111abbb(,,,,)MoNbTaV1bW1byyyMoNbTayyyyyVWMoNbTayyVW②③()Mo()()MoMo()()NbMo()()TaMo()()VMo()W11ab11ab11ab1a1b11ab105()Nb()()MoNb()()NbNb()()TaNb()()VNb()W11ab11ab11ab1a1b11ab()Ta()()MoTa()()NbTa()()TaTa()()VTa()W11ab11ab11ab1a1b11ab()()()()(VM11aboVN11abbV1a)(Ta11ba)(V)()V11ba(V)(W1b)()WM()()oWN()()bWT()()aWV()()WW()11ab11ab11ab1a1b11ab110图4BCC结构MoNbTaVW合金设计的计算途径Fig.4ThecalculationstepsoftheBCCstructureMoNbTaVWalloy115将生成焓ΔH转化为由端基化合物生成焓的权重迭加,其中权重系数为占位分数的乘积,如公式(4):11abEΔ=HyklyΔH(:)kl+ΔH(4)kAgNiZrlAgNiZr==,...,,...,,...,,...,-4-
中国科技论文在线http://www.paper.edu.cn120其中ΔH表示由室温稳定元素生成AB_B2型BCC结构相的端基化合物的生成焓,(:)klEEΔH为生成过剩焓,计算较为复杂,本文忽略,ΔS生成过剩熵也较为复杂,本文同样忽略。对于ΔH,可采用基于密度函数理论的第一性原理总能计算方法来计算,即:(:)kl11Δ=−HEEE−(5)()kl::totkl()totk()totl()22125由VASP计算得到端基化合物的总能后,利用公式(5)求得端基化合物的生成焓ΔH,(k:l)其中E为端基化合物的总能,E和E是端基化合物中相应单质元素的总能。然tot(k:l)tot(k)tot(l)后将得到多元多相体系端基化合物的生成焓数据库,进一步写成热力学计算软件Thermo-Calc的格式化的热力学数据库。130生成熵∆S可以合理地简化为:1111aa11bbΔ=SSconf.=−RyEiiln()yE+yEiiln()yE(6)22EAii==gN,...,iZ,...,rEAgN,...,iZ,...,r将公式(4)和(6)方程代入公式(3)中,同样可以得到AB_B2型BCC结构相Gibbs自由能,135如公式(7):111ab1a1aΔ=GyklyΔH()kl:+RyEiiln()yE+kA==gN,...,iZ,...,rlAgN,...,iZ,...,r2EAi=gN,...,iZ,...,r(7)111bbyyEEln()2iiEAgNiZri=,...,,...,依据质量守恒和成分归一化约束条件,则有:140xE=1(8)iEi=1-5-
中国科技论文在线http://www.paper.edu.cn1ayEi=1Ei=1(9)y1b=1EiEi=11111aby+=yx(10)22EEiiEi结合AB_B2型BCC结构相有序-无序转变达到平衡时的热力学条件,可以建立出包含145合金成分、温度和占位分数等变量的多元偏微分方程组。求解偏微分方程组,即可得到占位分数与合金成分和温度的关系,同时也可以计算得到合金体系的热力学函数(∆G、∆H和∆S)。1.3多主元高熵合金热力学函数求解基于第一性原理总能计算结果,建立一个含有多个备选相的格式化的端基化合物的生成150焓热力学数据库,(本研究组实际上建立了包含50个金属元素,FCC、BCC和Laves相这三种类型相的所有端基化合的生成焓热力学数据库)。进一步运用Thermo-Calc软件包,计算高熵合金体系的在不同温度下的热力学函数值ΔG,ΔH和ΔS值,以及在不同温度下的相组成,不同元素在各相亚点阵上的占位分数。进一步建立超晶胞,亚点阵上元素比例按占位分数计量,元素在亚点阵上随机占位,从而获得晶体中的原子分布构型,写成第一性原155理计算软件包VASP所需要的格式化的几何文件POSCAR,在高性能计算机上执行计算,就可以预测晶胞参数、总能和各种基本物理、力学性质。2结果与讨论忽略多元合金可能含有少量其它复杂金属间化合物相存在,考虑合金主要存在的FCC、BCC相以及可能存在的金属间化合物Laves相,利用包含有AB3_L12、AB_B2和AB2_C15160Laves三相热力学数据库,计算出合金在不同温度下的热力学函数和各种元素在不同亚晶格的占位分数。2.1MoNbTaVW合金的占位模拟表1列出了873K到2223K不同温度下,MoNbTaVW热力学函数和精细相结构计算模拟结果。MoNbTaVW合金在各个温度下的相结构均为BCC相,不同温度下,MoNbTaVW165的占位细节如下:在873K温度下(Mo0.399Nb0.054Ta0.067V0.107W0.374)1a(Mo0.001Nb0.346Ta0.333V0.293W0.026)3c在1623K温度下(Mo0.368Nb0.071Ta0.131V0.151W0.278)1a(Mo0.032Nb0.328Ta0.268V0.249W0.122)3c;在2223K温度下(Mo0.333Nb0.080Ta0.167V0.176W0.244)1a(Mo0.067Nb0.320Ta0.233V0.224W0.156)3c。在873K时,MoNbTaVW的构型熵10.43J/(mol∙K);在1623K的温度下,MoNbTaVW-6-
中国科技论文在线http://www.paper.edu.cn170的构型熵12.00J/(mol∙K);在1823K的温度下,MoNbTaVW的构型熵为12.30J/(mol∙K);在2023K的温度下,MoNbTaVW的构型熵为12.50J/(mol∙K);在2223K的温度下,MoNbTaVW的构型熵为12.60J/(mol∙K)。而按玻尔兹曼理想混合,各种元素在不同亚点阵上的占位分数为0.20,理想混合熵为13.36J/(mol∙K),显然,实际合金中,合金元素具有一定的占位倾向性,是偏离理想混合的。175表1MoNbTaVW热力学函数和精细相结构计算模拟结果Tab.1ThecalculationsresultofthethermodynamicfunctionsandfinephasestructureofMoNbTaVWTemperaturePhaseSiteoccupyingfractionΔGΔHΔSkJ/molkJ/molJ/(mol∙K)Name.Elem.Comp.1a1b2223KBCCMo0.200.3330.067-31.89-3.9112.60Nb0.200.0800.320Ta0.200.1680.233V0.200.1760.224W0.200.2440.1561623KBCCMo0.200.3680.032-24.47-4.9312.00Nb0.200.0710.329Ta0.200.1320.268V0.200.1510.249W0.200.2780.122873KBCCMo0.200.3990.001-15.98-6.8810.43Nb0.200.0540.346Ta0.200.0670.333V0.200.1070.293W0.200.3740.026图5是MoNbTaVW的占位分数随温度的关系图。随着温度逐渐升高,Mo和W在1a位置的占位倾向性逐渐下降,在1b位置的占位倾向性逐渐上升;Nb随温度的变化浮动较小,180说明Nb在1b位置倾向性更大且比较稳定;在2400K时,W、Ta和V在1a位置和1b位置的占位倾向性渐渐相近;在800K时,W和Mo更倾向于占据在1a位置,而Nb、Ta和V更倾向于占据在1b位置;Ta和V随温度升高,在1a位置的占位倾向性逐渐上升,在1b位置的占位倾向性逐渐下降。-7-
中国科技论文在线http://www.paper.edu.cn0.40Mo,1aNb,1a0.35Ta,1aV,1a0.30W,1aMo,1b0.25Nb,1bTa,1b0.20V,1b0.15W,1b0.10Siteoccupyingfraction0.050.006009001200150018002100240027003000Temperature,K185图5MoNbTaVW的占位分数与温度的关系Fig.5TherelationshipbetweensiteoccupyingfractionsandtemperatureofMoNbTaVW2.2MoNbTaVW合金的热力学亚晶格原子占位构型基于MoNbTaVW多主元高熵合金在不同温度下的占位分数,建立4×4×4的超晶胞方法,包含128个原子,且假设原子在每种单点阵上随机占位,建立原子排列的晶体几何构型,190并写出VASP软件包专用的格式化的POSCAR文件,并用VESTA软件可视化,建立原子的亚晶格占位模型。利用VASP软件,即可进一步预测合金相的晶胞体积,总能和力学性质。图6为MoNbTaVW在873K下的热力学亚晶格占位构型示意图;图7为MoNbTaVW在2223K下的热力学亚晶格占位模型示意图。可以清晰看出,在2223K时,蓝色的Mo和灰色的W占位有序,更倾向于占据1a点阵位置,黄色的Ta和绿色的Nb占位有序,更倾195向于占据1b点阵位置,而红色的V在两种亚晶格位置的占位比较均匀,趋于无序,与原子占位分数信息相吻合。图6MoNbTaVW在873K下的热力学亚晶格占位构型示意图200Fig.6ThermodynamicssublatticeoccupancyconfigurationofMoNbTaVWat873K-8-
中国科技论文在线http://www.paper.edu.cn图7MoNbTaVW在2223K下的热力学亚晶格占位构型示意图Fig.7ThermodynamicssublatticeoccupancyconfigurationofMoNbTaVWat2223K2052.3MoNbTaVW合金的固体状态方程曲线拟合运用VASP软件包计算得到不同体积下的总能,对Etot-V数据用Origin软件进行曲线拟合,拟合模型为三阶Birch-Murnaghan固体状态方程,如公式(11)所示,329VB22222200333E()V=E+V3V−1B"+V3V−16−4V3V0160000210(11)方程(11)含中包含有晶体的平衡能量(E0)、平衡体积(V0)、体弹性摸量(B0)及它对体积的一阶导数(B0")等结构、能量和物理力学性质,在材料设计中具有重要地位。3图8为MoNbTaVW在873K下的E-V曲线,超胞的平衡体积为2072.74Å,体积弹性215模量B0=P3×160.2189GPa=223.550GPa;图9为MoNbTaVW在2223K下的E-V曲线,超胞3的平衡体积为2078.90Å,体积弹性模量B0=P3×160.2189GPa=223.922GPa。可以看出,高熵合金的结构和体积弹性模量具有高温稳定性特征。-1404.0873KData:MoNbTaVW_873K-1404.5Chi^2=8.7057E-6-1405.0R^2=1P1-1407.60844±0.00162-1405.5P22072.74194±0.06891P31.39528±0.00197P43.83915±0.1479-1406.0,eV/128atoms-1406.5totE-1407.0-1407.5-1408.0198020002020204020602080210021202140216021803Volumeof4x4x4supercell,Angstrom图8MoNbTaVW超胞在873K下的E-V曲线220Fig.8TheE-VcurveofMoNbTaVWsupercellat873K-9-
中国科技论文在线http://www.paper.edu.cn-1405.52223KData:MoNbTaVW_2223K-1406.0Chi^2=3.0869E-6-1406.5R^2=1P1-1408.77633±0.00095P22078.90462±0.04234-1407.0P31.3976±0.00099P44.01313±0.08706-1407.5,eV/128atomstot-1408.0E-1408.5-1409.0198020002020204020602080210021202140216021803Volumeof4x4x4supercell,Angstrom图9MoNbTaVW超胞在2223K下的E-V曲线Fig.9TheE-VcurveofMoNbTaVWsupercellat2223K为了更加精确地计算弹性系数,弹性模量,泊松比,剪切模量,需要探索更加合理的方225法,比如应力-应变能方法,对高熵合金精细结构与力学性能之间的关系进行深入研究。3结论本文结合热力学亚晶格模型和量子化学第一性原理计算方法,以NiAl为原型的AB_B2(BCC)结构、Cu3Au为原型的AB3_L12(FCC)结构,以及以MgCu2为原型的Laves相结构的热力学双亚晶格模型,利用Thermo-Calc软件计算了MoNiTaVW合金的相结构、热230力学函数值和占位分数。基于占位分数,,运用单点阵上原子随机占位模式,建立原子排列的超晶胞原子分布构型模型,写出格式化的POSCAR文件,进一步预测了合金相的性能。MoNbTaVW合金在873~2223K的温度范围内由均BCC结构相组成,不同的合金元素占位不相同。Mn和Ta原子在FCC中占位有序,基本占据1a亚晶格位置;V和Nb原子在FCC中占位有序,基本占据1b亚晶格位置。而W原子在两种亚晶格位置的占位分数相差235比另外两种元素的较小,基本趋于无序。MoNbTaVW合金在873K、1623K和2223K温度下,4×4×4超晶胞平衡体积约为2075,体积弹性模量约为223GPa,表现出高温稳定性。[参考文献](References)[1]YehJW,ChenSK,LinSJ.Nano-structuredhighentropyalloyswithmultipleprincipalelements:novelalloy240designconceptsandoutcomes.AdvancedEngineeringMaterials[J].2004,6(5):299-303.[2]MiracleD.CriticalAssessment14:Highentropyalloysandtheirdevelopmentasstructuralmaterials[J].MaterialsScienceandTechnology,2015,31:1142-1147.[3]SenkovO,MillerJ,MiracleD,etal,Acceleratedexplorationofmulti-principalelementalloyswithsolidsolutionphases[J].NatureCommunication,2015,6:6529.245[4]ZhangY,ZhouYJ,LinJP,etal.Solid-solutionphaseformationrulesformulti-componentalloys[J].AdvancedEngineeringMaterials,2008,10(6):534-538.-10-
中国科技论文在线http://www.paper.edu.cn[5]WangFJ,ZhangY,ChenGL.Atomicpackingefficiencyandphasetransitioninahighentropyalloy[J].JournalofAlloysandCompounds,2009,478:321-324.[6]KingD.J.M,MiddleburghS.C,McGregorA.G,etal.Predictingtheformationandstabilityofsinglephase250high-entropyalloys[J].ActaMaterials,2016,104:172-179.[7]HuZH,ZhanYZ,ZhangGH,etal.EffectofrareearthYadditiononthemicrostructureandmechanicalpropertiesofhighentropyAlCoCrCuNiTialloys[J].MaterialsandDesign,2010,31:1599-1602.[8]杨晓宁,邓伟林,黄晓波,等.高熵合金制备方法进展[J].热加工工艺,2014,(22):30-33.[9]LiAM,ZhangXY.ThermodynamicanalysisofthesimplemicrostructureofAlCrFeNiCuhigh-entropyalloy255withmulti-principalelements[J].ActaMetallurgicaSinica,2009,22(3):219-224.[10]TroparevskyMC,MorrisJR,KentPRC,etal.CriteriaforPredictingtheFormationofSingle-PhaseHigh-EntropyAlloys[J].PhysicalReviewX,2015,5(1):011041.[11]LuZP,WangH,ChenMW,etal.Anassessmentonthefuturedevelopmentofhigh-entropyalloys:Summaryfromarecentworkshop[J].Intermetallics,2015,66:67-76.260[12]SenkovON,WilksGB,MiracleDB,etal.Refractoryhigh-entropyalloys[J].Intermetallics,2010,18(9):1758-1765.[13]GaoMC,CarneyCS,DoğanÖN,etal.DesignofRefractoryHigh-EntropyAlloys[J].JOM,2015,67(11):2653-2669.[14]SenkovON,ScottJM,SenkovaSV,etal.Microstructureandelevatedtemperaturepropertiesofarefractory265TaNbHfZrTialloy[J].JournalofMaterialsScience,2012,47(9):4062-4074.[15]GaoMC,YehJW,LiawPK,ZhangY,High-EntropyAlloys:FundamentalsandApplications[M],Springer,2016.[16]ZhangC,LinM,WuBetal.ExplorethePossibilityofFormingfccHighEntropyAlloysinEqual-AtomicSystemsCoFeMnNiMandCoFeMnNiSmM[J].JournalofShanghaiJiaotongUniversity(Science),2011,27016(2):173-179.-11-'
您可能关注的文档
- 基于德温特专利共现网络的石墨烯核心技术变迁趋势分析.pdf
- 基于正交试验分析熔融沉积成型精度因素.pdf
- 基于电流新息的改进配电网故障定位算法.pdf
- 基于稀疏约束的人体全身运动合成方法.pdf
- 基于网络结构和流量特征相似性的僵尸网络检测方法.pdf
- 基于虚拟化的windows驱动程序动态分析方法.pdf
- 基于视觉词和海明距离优化机制的相似图片检索系统的研究.pdf
- 基于雷达数据的高炉料层分布研究.pdf
- 基层扶贫资源分配的政府行为分析——以国家贫困县X县为例.pdf
- 多取代四氢-β-咔啉类衍生物的合成研究.pdf
- 大鼠灌胃蒙药复方阿拉坦-5后诃子酚性成分的药代动力学研究.pdf
- 套利活动对香港离岸人民币存款市场的影响研究.pdf
- 安徽巢北地区栖霞组臭灰岩段黄铁矿研究--形态及分布特征.pdf
- 家蚕BmPDCD2基因功能研究.pdf
- 岩溶地表水生系统不同季节的水化学昼夜变化特征及碳汇效应的研究--以重庆丰都雪玉洞流域地下水补给的水池为例.pdf
- 广东省碳排放与经济增长脱钩关系实证分析.pdf
- 微囊藻毒素合成酶McyG的N端结构域的结构研究.pdf
- 快充和超长稳定的高度互联Cu-Si合金纳米管锂离子电池阳极材料.pdf
相关文档
- 施工规范CECS140-2002给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程
- 施工规范CECS141-2002给水排水工程埋地钢管管道结构设计规程
- 施工规范CECS142-2002给水排水工程埋地铸铁管管道结构设计规程
- 施工规范CECS143-2002给水排水工程埋地预制混凝土圆形管管道结构设计规程
- 施工规范CECS145-2002给水排水工程埋地矩形管管道结构设计规程
- 施工规范CECS190-2005给水排水工程埋地玻璃纤维增强塑料夹砂管管道结构设计规程
- cecs 140:2002 给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程(含条文说明)
- cecs 141:2002 给水排水工程埋地钢管管道结构设计规程 条文说明
- cecs 140:2002 给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程 条文说明
- cecs 142:2002 给水排水工程埋地铸铁管管道结构设计规程 条文说明