

- 377.60 KB
- 2022-04-22 13:43:32 发布

- 1、本文档共5页,可阅读全部内容。
- 2、本文档内容版权归属内容提供方,所产生的收益全部归内容提供方所有。如果您对本文有版权争议,可选择认领,认领后既往收益都归您。
- 3、本文档由用户上传,本站不保证质量和数量令人满意,可能有诸多瑕疵,付费之前,请仔细先通过免费阅读内容等途径辨别内容交易风险。如存在严重挂羊头卖狗肉之情形,可联系本站下载客服投诉处理。
- 文档侵权举报电话:19940600175。
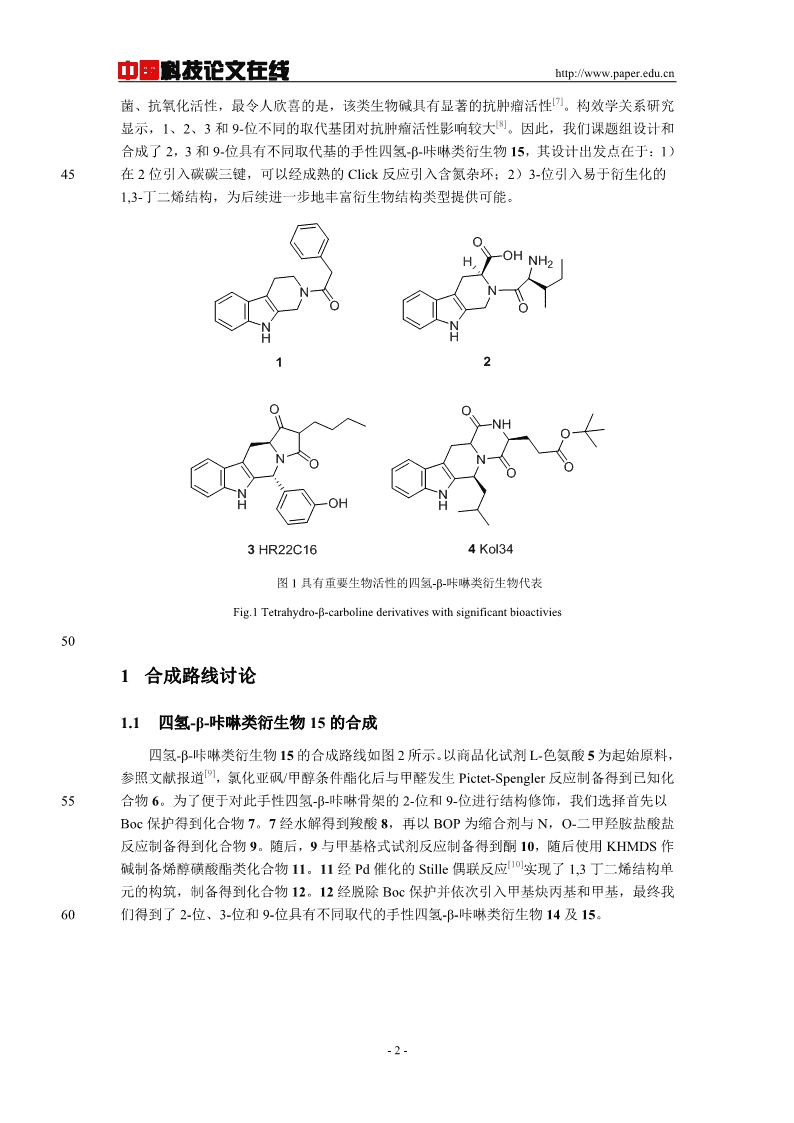
'中国科技论文在线http://www.paper.edu.cn#多取代四氢-β-咔啉类衍生物的合成研究**董佳威,张敏(重庆大学药学院,重庆401331)5摘要:β-咔啉类生物碱是一类重要的吲哚类生物碱,该类生物碱因为其在自然界中的广泛分布以及生物学活性的多样性而受到化学工作者们的广泛关注。药理学研究表明,β-咔啉类生物碱具有显著的抗肿瘤活性,且随着其取代基类型和位置的不同而有所改变。本文报道了一类从市售可得的L-色氨酸出发以Pictet-Spengler反应、Stille偶联等为关键反应高效制备102,3,9-位取代的四氢-β-咔啉类衍生物的方法。该方法为后续的β-咔啉类生物碱衍生物的生物活性研究奠定了坚实的基础。关键词:四氢-β-咔啉;Pictet-Spengler反应;吲哚生物碱;抗肿瘤活性中图分类号:O625.61+115StudyontheSynthesisofPolysubstitutedTetrahydro-β-carbolineDerivativesDONGJiawei,ZHANGMin(SchoolofPharmaceuticalSciences,ChongqingUniversity,Chongqing401331)Abstract:Theβ-carbolinealkaloidisanimportanttypeofindolealkaloids,andhasattracted20increasinginterestofsyntheticchemists,owingtoitswidedistributioninthenatureaswellasvarioussignificantbiologicalactivities.Previouspharmacologicalinvestigationshaveshownthatβ-carbolinealkaloidshaveremarkableantitumoractivities,whichvarywiththedifferentsubstitutions.Herein,wereportasynthesisstudyofchiral2,3,9-trisubstituted-1,2,3,4-tetrahydro-β-carbolinederivativeswithcommericallyavailableL-tryptophanasthestartingmaterialsandwithPictet-Spenglerreactionand25Stillecouplingasthekeysteps.Thisstudypavethebaseforthefurtherbiologicalstudiesofβ-carbolinealkaloidderivatives.Keywords:tetrahydro-β-carboline;Pictet-Spenglerreaction;indolealkaloids;antitumoractivity300引言[1]咔啉类生物碱是吲哚生物碱的一大家族,它包含一个相同的吡啶[3,4-b]并吲哚环的三元环骨架结构,依照氮原子在吡啶环中的不同相对位置,咔啉类生物碱被分为α、β及γ三大类,再根据吡啶环的不同饱和度,β-咔啉类生物碱又被进一步区分为β-咔啉,二氢-β-咔啉和四氢-β-咔啉。35含有β-咔啉类生物碱的植物长期被作为药物使用:生长于非洲东北部的一种蒺藜科植物骆驼蓬被当地人当做传统调经药和堕胎药;在我国西北部地区,骆驼蓬的种子提取物被应[2]用于治疗疟疾以及消化道癌症。近年来,越来越多的具有各样生物活性的咔啉类生物碱相[3,4]继被从不同种陆生植物、海洋微生物、昆虫以及哺乳动物的体液组织中分离提取得到。在这些结构各异的咔啉类化合物中,四氢-β-咔啉骨架因其在活性天然产物以及合成化合物[5,6]40中的广泛存在,吸引了众多化学工作者的关注,并对其进行了深入研究,我们简单列举含该骨架结构化合物于图1。研究表明,β-咔啉类生物碱不仅具有较好的抗炎、抗病毒、抗基金项目:重庆大学大型仪器设备开放基金(201512150100和201512150099)作者简介:董佳威(1993-),男,在读硕士生,药物化学通信联系人:张敏(1982-),研究员、博导,天然产物全合成、药物化学.E-mail:minzhang@cqu.edu.cn-1-
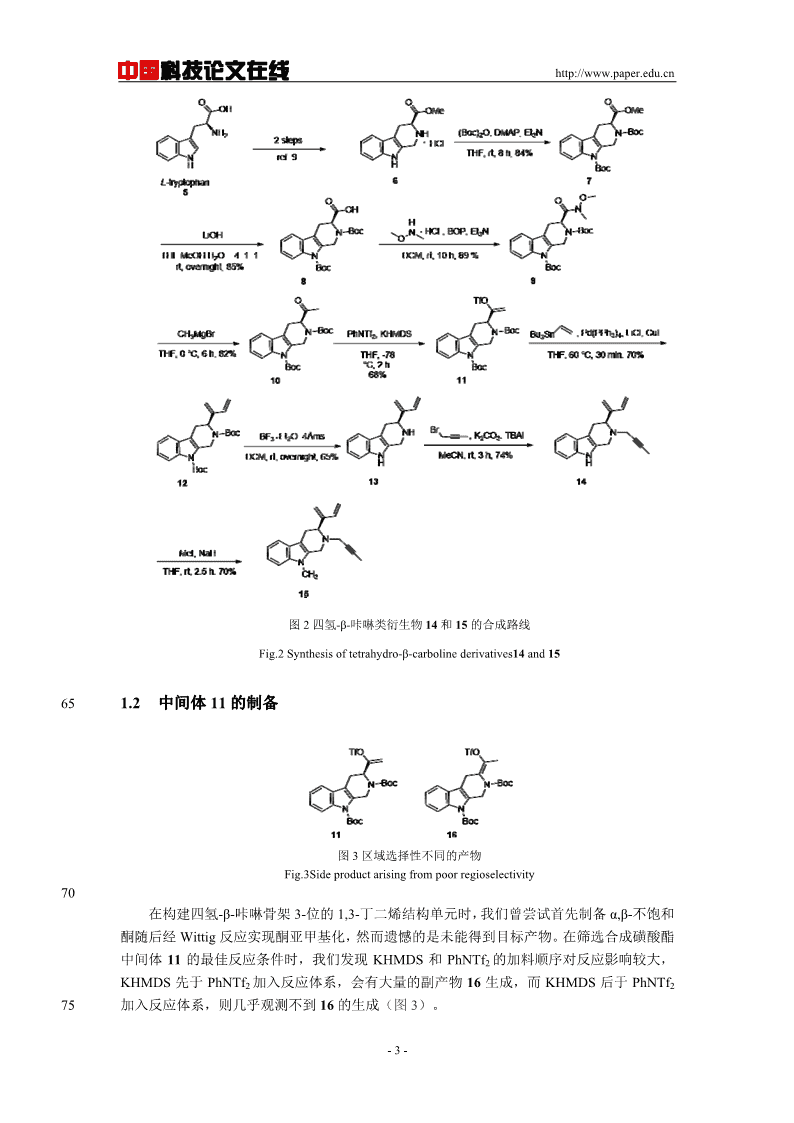
中国科技论文在线http://www.paper.edu.cn[7]菌、抗氧化活性,最令人欣喜的是,该类生物碱具有显著的抗肿瘤活性。构效学关系研究[8]显示,1、2、3和9-位不同的取代基团对抗肿瘤活性影响较大。因此,我们课题组设计和合成了2,3和9-位具有不同取代基的手性四氢-β-咔啉类衍生物15,其设计出发点在于:1)45在2位引入碳碳三键,可以经成熟的Click反应引入含氮杂环;2)3-位引入易于衍生化的1,3-丁二烯结构,为后续进一步地丰富衍生物结构类型提供可能。OOHHNH2NNOONNHH12OONHONNOOONNHOHH3HR22C164Kol34图1具有重要生物活性的四氢-β-咔啉类衍生物代表Fig.1Tetrahydro-β-carbolinederivativeswithsignificantbioactivies501合成路线讨论1.1四氢-β-咔啉类衍生物15的合成四氢-β-咔啉类衍生物15的合成路线如图2所示。以商品化试剂L-色氨酸5为起始原料,[9]参照文献报道,氯化亚砜/甲醇条件酯化后与甲醛发生Pictet-Spengler反应制备得到已知化55合物6。为了便于对此手性四氢-β-咔啉骨架的2-位和9-位进行结构修饰,我们选择首先以Boc保护得到化合物7。7经水解得到羧酸8,再以BOP为缩合剂与N,O-二甲羟胺盐酸盐反应制备得到化合物9。随后,9与甲基格式试剂反应制备得到酮10,随后使用KHMDS作[10]碱制备烯醇磺酸酯类化合物11。11经Pd催化的Stille偶联反应实现了1,3丁二烯结构单元的构筑,制备得到化合物12。12经脱除Boc保护并依次引入甲基炔丙基和甲基,最终我60们得到了2-位、3-位和9-位具有不同取代的手性四氢-β-咔啉类衍生物14及15。-2-
中国科技论文在线http://www.paper.edu.cn图2四氢-β-咔啉类衍生物14和15的合成路线Fig.2Synthesisoftetrahydro-β-carbolinederivatives14and15651.2中间体11的制备图3区域选择性不同的产物Fig.3Sideproductarisingfrompoorregioselectivity70在构建四氢-β-咔啉骨架3-位的1,3-丁二烯结构单元时,我们曾尝试首先制备α,β-不饱和酮随后经Wittig反应实现酮亚甲基化,然而遗憾的是未能得到目标产物。在筛选合成磺酸酯中间体11的最佳反应条件时,我们发现KHMDS和PhNTf2的加料顺序对反应影响较大,KHMDS先于PhNTf2加入反应体系,会有大量的副产物16生成,而KHMDS后于PhNTf275加入反应体系,则几乎观测不到16的生成(图3)。-3-
中国科技论文在线http://www.paper.edu.cn2实验部分2.1实验试剂与仪器本文所使用的的试剂购买自百灵威、安耐吉、阿达玛斯、泰坦科技、科龙化工等公司,均为A.R.或C.P.级。本文中二氯甲烷、乙腈采用回流除水干燥,干燥剂为氢化钙;甲醇用80镁条/碘体系进行干燥处理;四氢呋喃使用钠/二苯甲酮干燥。本文的反应均在氮气保护下进行,产物用柱层色谱分离(硅胶为青岛海洋化工厂生产),薄层板的显色采用紫外分析仪和碘、高锰酸钾、磷钼酸等显色剂。本文所有化合物核磁共振谱均由Agilent(400MHzDD2)型核磁共振波谱仪测定。852.2实验方法[9]合成路线如图2所示,化合物6参照文献方法合成。2.2.1化合物7的制备将化合物6(1.70g,6.37mmol)置于一个干燥洁净的50mL圆底烧瓶中,置换氮气三次,90加入干燥四氢呋喃(10.0mL)冷却至0℃,缓慢加入三乙胺(4.4ml,31.87mmol),继续加入4-二甲氨基吡啶(156mg,1.27mmol),冰浴下反应15min后,缓慢加入Boc酸酐(4.70g,21.53mmol),升至室温下反应约8小时后,TLC监测原料消失。反应液直接减压浓缩,粗产品经硅胶柱层析(石油醚:乙酸乙酯=15:1)分离纯化得到白色固体化合物7(2.31g,产率=84%)。1HNMR(400MHz,CDCl3)δ8.22-8.18(m,1H),7.43(d,J=7.3Hz,1H),7.31-7.22(m,2H),955.43(d,J=6.2Hz,1H),5.21-5.07(m,1H),4.65-4.52(m,1H),3.64(s,3H),3.38(d,J=16.1Hz,1H),3.06-2.99(m,1H),1.69(s,9H),1.55(s,9H)ppm.2.2.2化合物9的制备将经化合物7简单水解得到的的化合物8(5.68g,13.64mmol)置于一个干燥洁净的100100mL圆底烧瓶中,加入BOP缩合剂(10.86g,24.55mmol),N,O-二甲羟胺盐酸盐(3.33g,34.10mmol),置换氮气三次,于反应瓶中加入干燥的二氯甲烷(25.0mL),冰浴下缓慢滴加三乙胺(9.5mL,68.19mmol),反应液升至室温反应约10h,TLC监测原料消失。于冰浴下加入饱和氯化铵(15.0mL)淬灭反应,再加入二氯甲烷(20.0mL),分离出有机层,水相用二氯甲烷(20.0mL×3)萃取,合并有机相,用饱和食盐水(20.0mL)洗涤,有机相用无水硫酸钠干燥,过滤后105减压浓缩,粗产品经硅胶柱层析(石油醚:乙酸乙酯=5:1)分离纯化得白色固体化合物9(5.581g,产率=89%)。HNMR(400MHz,CDCl3)δ8.23(d,J=8.3Hz,1H),7.38(d,J=7.5Hz,1H),7.28-7.19(m,2H),5.66(d,J=4.5Hz,1H),5.25-5.07(m,1H),4.97-4.81(m,1H),3.86(s,3H),3.12(s,5H),1.68(s,9H),1.53(s,9H)ppm.-4-
中国科技论文在线http://www.paper.edu.cn1102.2.3化合物10的制备将化合物9(567.3mg,1.23mmol)置于一个干燥洁净的25mL圆底烧瓶中,置换氮气三次,加入干燥的四氢呋喃(10.0mL),于冰浴下缓慢滴加甲基溴化镁(3.0mL,3.00mmol),冰浴下反应约6h,TLC监测原料消失。于冰浴下加入饱和氯化铵(10.0mL)淬灭反应,再加入乙酸乙酯(10.0mL),分离出有机层,水相用乙酸乙酯(10.0mL×3)萃取,合并有机相,用饱115和食盐水(10.0mL)洗涤,有机相用无水硫酸钠干燥,过滤后减压浓缩,粗产品经硅胶柱层析1(石油醚:乙酸乙酯=8:1)分离纯化得浅黄色油状化合物10(420.0mg,产率=82%)。HNMR(400MHz,CDCl3)δ8.18(d,J=8.3Hz,1H),7.44(d,J=7.4Hz,1H),7.30-7.22(m,2H),5.24(d,J=6.7Hz,1H),5.16-5.10(m,1H),4.57(d,J=18.3Hz,1H),3.34(t,J=16.1Hz,1H),3.00-2.91(m,1H),2.15(s,3H),1.68(s,9H),1.54(s,9H)ppm.1202.2.4化合物11的制备将化合物10(988.8mg,2.39mmol)置于一个干燥洁净的50mL圆底烧瓶中,加入N-苯基双(三氟甲烷磺酸亚胺)(1.79g,5.01mmol),置换氮气三次,加入干燥的四氢呋喃(20.0mL),-78℃下逐滴滴加双(三甲基硅烷基)氨基钾(6.9mL,6.92mmol),反应2h,TLC监测原料消失。125于-78℃下加入饱和氯化铵(10.0mL)淬灭反应,再加入乙酸乙酯(10.0mL),分离出有机层,水相用乙酸乙酯(10.0mL×3)萃取,合并有机相,用饱和食盐水(10.0mL)洗涤,有机相用无水硫酸钠干燥,过滤后减压浓缩,粗产品经硅胶柱层析(石油醚:乙酸乙酯=20:1)分离纯1化得黄色油状化合物11(886.6mg,产率=68%)。HNMR(400MHz,CDCl3)δ8.17(s,1H),7.43(d,J=7.5Hz,1H),7.33-7.24(m,2H),5.60-5.21(m,3H),4.90(d,J=9.7Hz,1H),4.25130(q,J=19.2Hz,1H),3.14-3.01(m,2H),1.69(s,9H),1.52(s,9H)ppm.2.2.5化合物12的制备将化合物11(582.9mg,1.07mmol)置于一个干燥洁净的50mL圆底烧瓶中,依次加入四三苯基膦钯(247.0mg,0.21mmol),无水氯化锂(362.0mg,8.53mmol),氯化亚铜(824.0mg,1358.32mmol),置换氮气三次,常温下加入干燥的四氢呋喃(10.0mL),冰浴下加入乙烯基三正丁基锡(0.63mL,2.14mmol),移至60℃反应约30min,TLC监测原料消失。反应液经硅藻土过滤并减压浓缩,粗产品经硅胶柱层析(石油醚:乙酸乙酯=20:1)分离纯化得黄色油状1化合物12(316.6mg,产率=70%)。HNMR(400MHz,CDCl3)δ8.18(d,J=8.0Hz,1H),7.44(d,J=7.4Hz,1H),7.30-7.22(m,2H),6.30(dd,J=17.7,11.1Hz,1H),5.56-5.10(m,5H),1404.81(s,1H),4.00(d,J=18.1Hz,1H),3.09-2.95(m,2H),1.66(s,9H),1.50(s,9H)ppm.2.2.6化合物13的制备将化合物12(320.0mg,0.75mmol)置于一个干燥洁净的25mL圆底烧瓶中,加入少许4Ams,置换氮气三次,加入干燥的二氯甲烷(5.0mL),于0℃下缓慢加入三氟化硼乙醚(1.6mL,6.03145mmol),升至室温反应过夜,TCL监测原料消失。于冰浴下加入饱和碳酸氢钠溶液直至不产生气泡,加入二氯甲烷(10.0mL),分离出有机层,水相用二氯甲烷(10.0mL×3)萃取,合并有机相,用饱和食盐水(10.0mL)洗涤,有机相用无水硫酸钠干燥,过滤后减压浓缩,粗产品经硅胶柱层析(二氯甲烷:甲醇=25:1)分离纯化得浅黄色油状化合物13(109.6mg,产率-5-
中国科技论文在线http://www.paper.edu.cn1=65%)。HNMR(400MHz,CDCl3)δ7.81(s,1H),7.47(d,J=5.2Hz,1H),7.31(d,J=5.6Hz,1150H),7.15(t,J=5.1Hz,1H),7.09(t,J=5.0Hz,1H),6.46(dd,J=11.8,11.2Hz,1H),5.42(d,J=11.7Hz,1H),5.32(s,1H),5.23(s,1H),5.17(d,J=7.4Hz,1H),4.22(d,J=10.4Hz,1H),4.13(d,J=10.2Hz,1H),3.84(dd,J=6.9,2.6Hz,1H),3.03(d,J=10.3Hz,1H),2.70(t,J=8.9Hz,1H)ppm.1552.2.7化合物14的制备将化合物13(143.6mg,0.64mmol)置于一个干燥洁净的反应试管中,依次加入无水碳酸钾(265.5mg,1.92mmol),四丁基碘化铵(25.8mg,0.07mmol),置换氮气三次,加入干燥的乙腈(2mL),于冰浴下滴加1-溴-2-丁炔(0.14mL,1.6mmol),升至室温反应约3h,TLC监测原料消失。于冰浴下加入饱和氯化铵(2.0mL)淬灭反应,再加入乙酸乙酯(2.0mL),分离出有机160层,水相用乙酸乙酯(2.0mL×3)萃取,合并有机相,用饱和食盐水(2.0mL)洗涤,有机相用无水硫酸钠干燥,过滤后减压浓缩,粗产品经硅胶柱层析(石油醚:乙酸乙酯=8:1)分离纯1化得浅黄色油状化合物14(130.9mg,产率=74%)。HNMR(400MHz,CDCl3)δ7.76(s,1H),7.44(d,J=7.8Hz,1H),7.30(d,J=7.9Hz,1H),7.16-7.07(m,2H),6.44(dd,J=17.6,11.1Hz,1H),5.74(d,J=17.6Hz,1H),5.33(s,1H),5.24(s,1H),5.16(d,J=11.1Hz,1H),3.97(q,J=16515.2Hz,2H),3.68-3.64(m,1H),3.46(q,J=17.3Hz,2H),3.02(t,J=12.7Hz,1H),2.85(d,J=15.7Hz,1H),1.83(s,3H)ppm.2.2.8化合物15的制备将化合物14(309.6mg,1.12mmol)置于一个干燥洁净的反应试管中,置换氮气三次,加170入干燥的四氢呋喃(2.0mL),冰浴下缓慢加入氢化钠(90.0mg,2.24mmol),0℃搅拌10min后,缓慢滴加碘甲烷(0.08mL,1.23mmol),升至室温反应约2.5h,TCL监测原料消失。于冰浴下加入饱和氯化铵(2.0mL)淬灭反应,再加入乙酸乙酯(2.0mL),分离出有机层,水相用乙酸乙酯(2.0mL×3)萃取,合并有机相,用饱和食盐水(2.0mL)洗涤,有机相用无水硫酸钠干燥,过滤后减压浓缩,粗产品经硅胶柱层析(石油醚:乙酸乙酯=20:1)分离纯化得黄1175色油状化合物15(227.5mg,产率=70%)。HNMR(400MHz,CDCl3)δ7.44(d,J=7.7Hz,1H),7.28-7.26(m,1H),7.17(t,J=7.6Hz,1H),7.07(t,J=7.4Hz,1H),6.43(dd,J=17.6,11.1Hz,1H),5.74(d,J=17.6Hz,1H),5.32(s,1H),5.24(s,1H),5.16(d,J=11.1Hz,1H),4.05(d,J=15.1Hz,1H),3.92(d,J=15.1Hz,1H),3.64(s,3H),3.64-3.43(m,3H),3.04(t,J=12.8Hz,1H),2.86(d,J=15.6Hz,1H),1.84(s,3H)ppm.1803结论以市售可得的L-色氨酸为起始原料,经酯化、Pictet-Spengler反应、水解、Stille偶联等11步反应,我们以较高收率成功制备了2,3和9-位取代的手性四氢-β-咔啉类衍生物14和15。由于14和15具有1,3-丁二烯和炔基等易于衍生化的基团,以此为原料易于制备更多样185性的四氢-β-咔啉类衍生物。14和15及其衍生物的生物活性及其构效学关系研究将是我们后续研究工作的重点。-6-
中国科技论文在线http://www.paper.edu.cn4致谢感谢重庆大学大型仪器设备开放基金(201512150100和201512150099)的资助。190[参考文献](References)[1]AllenJRF,HolmstedtBR.Thesimpleβ-carbolinealkaloids[J].Phytochemistry,1980,19(8):1573-1582.[2]ZengHM,ZhengRS,GuoYM,etal.CancersurvivalinChina,2003-2005:Apopulation-basedstudy[J].InternationalJournalofCancer,2015,136(8):1921-1930.[3]PfauW,SkogK.Exposuretoβ-carbolinenorharmanandharman[J].JournalofChromatographyB,2004,195802(1):115-126.[4]LiuJW,JiangXY,ZhaoM,etalAclassof3S-2-aminoacyltetrahydro-β-carboline-3-carboxylicacids:Theirfacilesynthesis,inhibitionforplateletactivation,andhighinvivoanti-thromboticpotency[J].JournalofMedicinalChemistry,2010,53(8):3106-3116.[5]CarbreraGM,SeldesAM.Abeta-carbolinealkaloidfromthesoftcorallignopsisspongiosum[J].Journalof200NaturalProducts,1999,62:759-760.[6]ZhengC,FangYZ,ChenYH.Synthesisandbiologicalevaluationofnoveltetrahydro-β-carbolinederivativesasanti-tumorgrowthandmetastasisagentsthroughinhibitingthetransforminggrowthfactor-βsignalingpathway[J].JournalofMedicinalChemistry,2014,57(3):600-612.[7]IshidaJ,WangHK,etal.Antitumoragents201.1cytotoxicityofharmineandβ-carbolineanalogs[J].205Bioorganic&MedicinalChemistryLetters,1999,9(23):3319-3324.[8]CaoRH,PengWL,WangZH,XuAL.β-carbolinealkaloids:biochemicalandpharmacologicolfunctions[J].CurrentMedicinalChemistry,2007,14(4):479-500.[9]EfremovIV,VajdosFF,BorzilleriKA,etal.Discoveryandoptimizationofanovelspiropyrrolidineinhibitorofβ-secretase(BACE1)throughfragment-baseddrugdesign[J].JournalofMedicinalChemistry,2012,55(21):2109069-9088.[10]HanXJ,StoltzBM,CoreyEJ.Cuprouschlorideacceleratedstillereactions.Ageneralandeffectivecouplingsystemforstericallycongestedsubstratesandforenantioselectivesynthesis[J].JournaloftheAmericanChemicalSociety,1999,121(33):7600-7605.-7-'
您可能关注的文档
- 基于正交试验分析熔融沉积成型精度因素.pdf
- 基于电流新息的改进配电网故障定位算法.pdf
- 基于稀疏约束的人体全身运动合成方法.pdf
- 基于网络结构和流量特征相似性的僵尸网络检测方法.pdf
- 基于虚拟化的windows驱动程序动态分析方法.pdf
- 基于视觉词和海明距离优化机制的相似图片检索系统的研究.pdf
- 基于雷达数据的高炉料层分布研究.pdf
- 基层扶贫资源分配的政府行为分析——以国家贫困县X县为例.pdf
- 多主元高熵合金MoNbTaVW中合金元素的占位行为.pdf
- 大鼠灌胃蒙药复方阿拉坦-5后诃子酚性成分的药代动力学研究.pdf
- 套利活动对香港离岸人民币存款市场的影响研究.pdf
- 安徽巢北地区栖霞组臭灰岩段黄铁矿研究--形态及分布特征.pdf
- 家蚕BmPDCD2基因功能研究.pdf
- 岩溶地表水生系统不同季节的水化学昼夜变化特征及碳汇效应的研究--以重庆丰都雪玉洞流域地下水补给的水池为例.pdf
- 广东省碳排放与经济增长脱钩关系实证分析.pdf
- 微囊藻毒素合成酶McyG的N端结构域的结构研究.pdf
- 快充和超长稳定的高度互联Cu-Si合金纳米管锂离子电池阳极材料.pdf
- 提高环境质量的财政基础.pdf
相关文档
- 施工规范CECS140-2002给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程
- 施工规范CECS141-2002给水排水工程埋地钢管管道结构设计规程
- 施工规范CECS142-2002给水排水工程埋地铸铁管管道结构设计规程
- 施工规范CECS143-2002给水排水工程埋地预制混凝土圆形管管道结构设计规程
- 施工规范CECS145-2002给水排水工程埋地矩形管管道结构设计规程
- 施工规范CECS190-2005给水排水工程埋地玻璃纤维增强塑料夹砂管管道结构设计规程
- cecs 140:2002 给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程(含条文说明)
- cecs 141:2002 给水排水工程埋地钢管管道结构设计规程 条文说明
- cecs 140:2002 给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程 条文说明
- cecs 142:2002 给水排水工程埋地铸铁管管道结构设计规程 条文说明