

- 995.64 KB
- 2022-04-22 13:42:43 发布


- 1、本文档共5页,可阅读全部内容。
- 2、本文档内容版权归属内容提供方,所产生的收益全部归内容提供方所有。如果您对本文有版权争议,可选择认领,认领后既往收益都归您。
- 3、本文档由用户上传,本站不保证质量和数量令人满意,可能有诸多瑕疵,付费之前,请仔细先通过免费阅读内容等途径辨别内容交易风险。如存在严重挂羊头卖狗肉之情形,可联系本站下载客服投诉处理。
- 文档侵权举报电话:19940600175。
'中国科技论文在线http://www.paper.edu.cn珠三角地区有机硝酸的模拟及其对臭氧生#成的化学影响分析**陈肖睿,王海潮,陆克定5(北京大学环境科学与工程学院,北京市100871)摘要:有机硝酸是大气污染过程中重要的二次污染物,与臭氧和气溶胶的生成密切相关。本研究以珠三角后花园地区2006年的综合观测数据为基础,基于盒子模型搭载RACM2机理对有机硝酸开展模拟,发现RACM2高估了日间有机硝酸生成速率,进而优化了RACM210机理中日间有机硝酸的生成模块,优化后有机硝酸的有效产率(αeff)在0.033左右;根据VOC-OH反应活性、有机硝酸产率和臭氧生成速率三者之间的关系,分析了有机硝酸的生成过程对臭氧化学的影响,结果显示VOC-OH反应活性与αeff呈正相关,在高VOC-OH反应活性区域臭氧的生成受到有机硝酸生成增强的明显抑制,P(Ox)随VOC-OH反应活性的升高而表现出先上升后下降的趋势。这一结果表明在通过削减VOCs来控制O3时,需要15进一步考虑有机硝酸的生成所带来的化学影响;即在珠三角光化学污染季削减臭氧时,需要优先考虑低分子量VOCs的削减。关键词:大气环境学;有机硝酸;臭氧;RACM2;珠三角中图分类号:X51120MedellingoforganicnitratesinPearlRiverDelta2006andtheimpactonozonechemsitryproductionChenXiaorui,WangHaichao,LuKeding(CollegeofEnvironmentSciencesandEngineering,PekingUniversity,Beijing100871)Abstract:Organicnitrateisanimportantkindofsecondarypollutantsintheatmosphereandclosely25relatedtotheproductionofozoneaswellasaerosol.ThispaperisbasedontheintensivecampaignatBackgardenofPRD,simulatingthechemistryproductionoforganicnitratesusingboxmodelwithRACM2.Themodeloverratedthedaytimeproductionsoweoptimizedtherelevantpartofmechnism,theeffectivebranchingratio(αeff)ofwhichreachingabout0.033.TheimpactoforganicnitratesonozoneproductionwereanalysizedaccordingtotherelationbetweenVOC-OHreactivity,effective30branchingratioandozoneproductionrates.TheresultsillustratedthattheincreaseofVOC-OHreactivityledtohigherαeff,howeverwascouterproductivetoozoneproductionathighlyreactivesituationduetoincreaseoforganicnitrateproduction.Itenlightenedthattheimpactoforganicnitrateonozoneproductionwasessentialforcontrollingozonepollution,whichgaveprioritytoreducingVOCsoflowmoleculeweightinPRD.35Keywords:Atmosphericenvironment;Organicnitrates;Ozone;RAMC2;PearlRiverDelta0引言有机硝酸(RONO2)是大气污染中重要的二次污染物,对区域乃至全球的大气氧化性以及基金项目:国家自然科学基金(批准号:41375124,21522701,91544225,41421064);中国科学院战略性先导科技专项项目(编号:XDB05010500);博士点基金(20130001120010);国家科技支撑计划(2014BAC21B01)作者简介:陈肖睿,男,主要研究方向:大气化学通信联系人:陆克定,男,研究员、博导,主要研究方向:大气化学.E-mail:k.lu@pku.edu.cn-1-
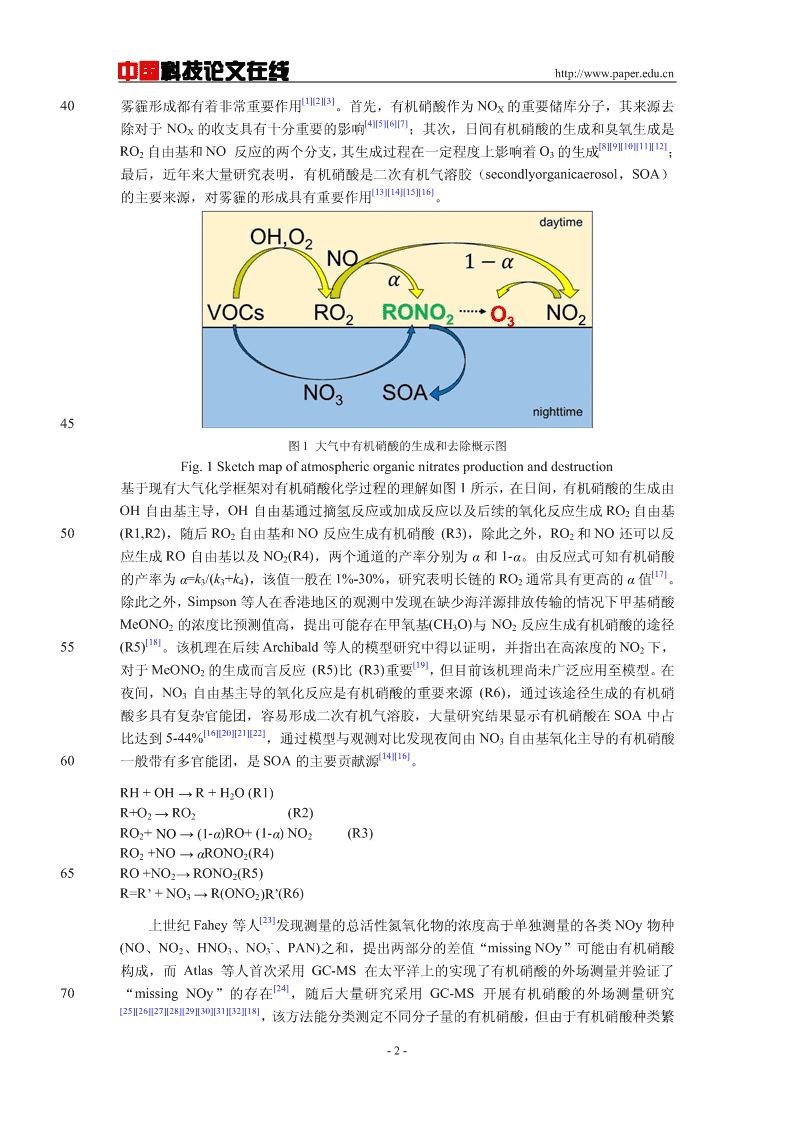
中国科技论文在线http://www.paper.edu.cn[1][2][3]40雾霾形成都有着非常重要作用。首先,有机硝酸作为NOX的重要储库分子,其来源去[4][5][6][7]除对于NOX的收支具有十分重要的影响;其次,日间有机硝酸的生成和臭氧生成是[8][9][10][11][12]RO2自由基和NO反应的两个分支,其生成过程在一定程度上影响着O3的生成;最后,近年来大量研究表明,有机硝酸是二次有机气溶胶(secondlyorganicaerosol,SOA)[13][14][15][16]的主要来源,对雾霾的形成具有重要作用。45图1大气中有机硝酸的生成和去除概示图Fig.1Sketchmapofatmosphericorganicnitratesproductionanddestruction基于现有大气化学框架对有机硝酸化学过程的理解如图1所示,在日间,有机硝酸的生成由OH自由基主导,OH自由基通过摘氢反应或加成反应以及后续的氧化反应生成RO2自由基50(R1,R2),随后RO2自由基和NO反应生成有机硝酸(R3),除此之外,RO2和NO还可以反应生成RO自由基以及NO2(R4),两个通道的产率分别为α和1-α。由反应式可知有机硝酸[17]的产率为α=k3/(k3+k4),该值一般在1%-30%,研究表明长链的RO2通常具有更高的α值。除此之外,Simpson等人在香港地区的观测中发现在缺少海洋源排放传输的情况下甲基硝酸MeONO2的浓度比预测值高,提出可能存在甲氧基(CH3O)与NO2反应生成有机硝酸的途径[18]55(R5)。该机理在后续Archibald等人的模型研究中得以证明,并指出在高浓度的NO2下,[19]对于MeONO2的生成而言反应(R5)比(R3)重要,但目前该机理尚未广泛应用至模型。在夜间,NO3自由基主导的氧化反应是有机硝酸的重要来源(R6),通过该途径生成的有机硝酸多具有复杂官能团,容易形成二次有机气溶胶,大量研究结果显示有机硝酸在SOA中占[16][20][21][22]比达到5-44%,通过模型与观测对比发现夜间由NO3自由基氧化主导的有机硝酸[14][16]60一般带有多官能团,是SOA的主要贡献源。RH+OH→R+H2O(R1)R+O2→RO2(R2)RO2+NO→(1-α)RO+(1-α)NO2(R3)RO2+NO→αRONO2(R4)65RO+NO2→RONO2(R5)R=R’+NO3→R(ONO2)R’(R6)[23]上世纪Fahey等人发现测量的总活性氮氧化物的浓度高于单独测量的各类NOy物种-(NO、NO2、HNO3、NO3、PAN)之和,提出两部分的差值“missingNOy”可能由有机硝酸构成,而Atlas等人首次采用GC-MS在太平洋上的实现了有机硝酸的外场测量并验证了[24]70“missingNOy”的存在,随后大量研究采用GC-MS开展有机硝酸的外场测量研究[25][26][27][28][29][30][31][32][18],该方法能分类测定不同分子量的有机硝酸,但由于有机硝酸种类繁-2-
中国科技论文在线http://www.paper.edu.cn多,目前尚且不能测量到完整种类的有机硝酸,且GC-MS的时间分辨率较低,对有机硝酸的浓度变化以及时空分布等信息的捕捉能力有限。近年来人们利用有机硝酸在高温下易分解的性质,开发了通过测量有机硝酸热解后生成的NO2来实现总有机硝酸的间接测量,包括75TD-LIF和TD-CRDS等,目前该类仪器已经能够满足总有机硝酸的实验室和外场观测的测[33][34][35][36][37][38][39][40][41]量。结合上述测量手段的总有机硝酸与单类有机硝酸观测有助于分析[42][11]有机硝酸的收支及其前体物的来源贡献,厘清有机硝酸的化学生成机制。尽管如此,受到测量技术的限制,目前基于有机硝酸外场测量的相关研究成果仍然比较缺乏。除了测量之外,模型模拟或数值计算也是探索有机硝酸化学机制以及环境影响的有效手[5][43][1][44][2][45]80段。已有研究表明RACM机理第二版本(RACM2)对于HOx自由基以及O3浓[46][47]度的模拟表现出很好的效果,但缺乏相关研究来评价该套机理中对有机硝酸的模拟,因此本研究以2006年珠三角后花园地区综合观测(PRIDE-PRD2006)的观测数据为基础,对RACM2机理中有机硝酸的化学机制进行探讨,比对模式模拟得到的有机硝酸的生成速率与理论计算值,并对RACM2中有机硝酸的产率α进行修正,得到混合气团有效αeff的变化范85围,并探讨了不同VOCs气团组成下有机硝酸的生成与臭氧生成的化学关系。1方法1.1观测活动概述珠三角地区位于中国广东省,是一个涵盖了广州、深圳、香港等大中型城市在内的工业区域和港口,其海陆空交通网络发达。该地区的区域快速城市化导致区域空气污染问题累积[48][49]90。2006年夏季开展的“珠三角地区空气质量区域综合观测实验”(PRIDE-PRD2006)致力于研究城市郊区环境下气相化学以及气溶胶形成过程,综合观测站点位于广州市西北方向60km(见图2)处的后花园超级站(23.5°N,113.03°E),地处水库周边,周围主要种植一些经济农用作物,如荔枝和花生。该超级站受广州城区的交通污染排放较小,受生物源排放影响较多,偶尔会有周边的生物质燃烧产生污染气团。观测实验是从2006年7月3日开始至3095日结束,为期近一个月。观测期间受亚热带气候影响,雨量丰富,期间经历了两次台风Bilis(7月15日-18日)和Kaemi(7月26日-29日)。观测期间的温度、湿度均较高(温度28-36℃、湿度60-90%),风速普遍低于2m/s,珠三角地区(尤其是广州市区)的污染气团在沿途充分老化,在此受体站点混合均匀。-3-
中国科技论文在线http://www.paper.edu.cn100图2观测点后花园超级站的位置,其中红点表示后花园地区,蓝点代表广州市中心地区Fig.2LocationofBackgardencampaignsite.ReddotshowsBackgardenandbluedotshowsGuangzhoudowntown1.2痕量气体的观测本次观测中主要的痕量气体包括NO,NO2,NOy,O3,HONO,VOCs等,相应的测量105仪器概况数据见表1,详细的仪器细节可参见Lou等(2010)。大部分痕量气体的采样高度约10米,HONO和气象数据的采样口高度约7米,两个采样口水平距离约为30m。光解速率1值测量手段和测量结果可参见Lu等(2012),包括j(OD)、j(NO2)、j(HONO)、j(HCHO)。各类痕量气体的日间和夜间平均浓度水平在表2中给出。表1观测的主要痕量气体及其测量手段110Tab.1InstrumentsforatmospherictracegasesmeasurementsduringthecampaignCompoundTechniquesTimeresolutionLOD(1σ)AccuracyNOChemiluminescence1min50pptv7%NO2PhotolyticalconverterandChemiluminescence1min170pptv13%NOyHeatedgoldconverterandChemiluminescence1min68pptv12%O3UVphotometry1min0.3ppbv5%HONOLong-pathabsorptionphotometry5min7pptv10%COIRphotometry1min4ppbv5%CH4Fourier-transforminfraredspectrometry10min0.5ppmv4%C2H6Canistersample-GC10s2pptv5%C2H4Canistersample-GC10s3pptv10%C3-C12Online-GC1h6–70pptv10%表2观测期间主要痕量气体的平均浓度水平Tab.2MeanvaluesofmeasuredtracegasesduringthecampaignMeanmixingratio(ppbv)TracegasDay(06:00-18:00)Night(18:00-06:00)NO4.033.18NO29.9017.0-4-
中国科技论文在线http://www.paper.edu.cnNOy15.423.9O335.115.9HONO0.520.97CO605725CH420892240aEthane1.51.5aEthene3.03.0Isoprene0.980.79bHC34.345.95bHC53.095.32bHC82.224.03bOLI0.250.50bOLT1.552.60bBEN1.572.36bTOL3.376.00a.设定为常数,由罐采样GC分析的结果估计得到,具体见1.3节文字部分;115b.RACM2中的归纳物质1.3模型与机理本文采用零维盒子模型搭载RACM2机理对本次珠三角观测中的有机硝酸进行模拟,模拟中不含气溶胶相的反应过程及水平和垂直扩散,物质浓度在盒子内均匀。边界约束条件包括O3、HONO、NO、NO2、CO、CH4、C3-C12的VOCs、光解速率、湿度、温度、压力,120其中C2H6和C2H4分别用固定值1.5ppbv和3.0ppbv约束(由罐采样结果估算得到,其浓度变化对有机硝酸浓度影响极小),H2的浓度则用固定值550ppbv约束。模型的时间分辨率统一为30min,同时模型对所有物质赋予了寿命为24h的干沉降过程防止一些OVOC的非正常累积。另外,模型对有机硝酸的归纳物质分别赋予了8h和7h的沉降过程,该假设被认[36][50][46][47]为是合理的。该模型的更多细节描述和准确性评估可参见之前的研究。125在RACM2中有机硝酸主要由ONIT和ISON两种归纳物质表示,其中ISON代表的是由异戊二烯氧化所得到的有机硝酸,而ONIT则囊括了其他VOC前体物所氧化得到的有机硝酸。虽然机理中仅将有机硝酸简单的归纳成上述两类物质,但对它们的生成机制仍有系统的描述。通过比对RACM2的归并机理计算得到的有机硝酸生成速率和利用观测得到的各类VOC计算出的理论生成速率,可以验证机理对有机硝酸生成机制描述的可靠性。1302.结果与讨论2.1NOz的缺失“missingNOz”(NOz=NOy-NOX)是指测量的总NOZ与分类测量的ΣNOZi之间的差值,而“missingNOy”是测量的总NOy与分类测量的ΣNOyi之间的差值。早期研究表明在中纬度内陆地区“missingNOy”占NOy之比能达到10-60%,且浓度呈现清晰的日变化以及季节变化规[51][52][53][54][55]135律,一般在夏季或午后较高,说明了该部分物质和光化学过程关系密切。Day等(2003)研究表明有机硝酸在NOy中占比显著,能够较好的和“missingNOy”吻合,因此认为“missingNOy”中主要成分是有机硝酸。由于在后花园地区NOx浓度较高(中值70%)导致“missingNOy”日变化趋势不显著,而研究“missingNOZ”可以排除高浓度NOx的影-5-
中国科技论文在线http://www.paper.edu.cn响,因此可以借助该参数来反映有机硝酸的日变化、占比等特征,等效于研究“missingNOy”。140本次观测中测量的NOzi包括HNO3、HONO、∑PNs(PAN+PPN),其各自的日均浓度变化如图3(a)所示,HNO3、HONO、∑PNs各占NOz的20%左右,剩余的40%即为“missingNOz”。N2O5在观测期间没有测量值,通过估算约占NOz的0.5%左右,若“missingNOz”全部由有机硝酸构成,那么珠三角地区的有机硝酸占NOZ的比例接近40%。表3总结了近年观测中有机硝酸在NOy或NOz中的占比,可以看到在有ΣRONO2的观测中,郊区的有机硝酸在NOy[37]145中占比相对城市地区较高,这是因为这些地区普遍处于城区的下风向,气团老化程度高。而在航测中由于飞机飞行高度通常超过混合层,NOx被滴定使得有机硝酸的占比较高。在总结的观测中,仅有美国德州和加州的两次观测有ΣRONO2/NOz的统计值,其最大占比不超过50%,且高占比的情况较多出现在冬季,夏季的ΣRONO2/NOz值变化范围在20-30%之[34]间。本次夏季观测地处北半球中低纬度,与上述两次观测所处纬度类似,150“missingNOz”/NOz的变化范围为0.2-90%,“missingNOz”/NOy最大值为56.7%,高值均超过历次的观测。若假设“missingNOz”主要由有机硝酸组成,则说明后花园地区有机硝酸可能在氮氧化物的收支中起到十分重要的作用。[5][12][26][27][31][34][36][37][42][50][56][57]表3城区、郊区及航测所得ΣRONO2/NOy与ΣRONO2/NOzTab.3ΣRONO2/NOyaswellasΣRONO2/NOzmeasuredaturban,suburbansitesandonaircraftsccLocationMeasuredOrganicNitratesΣRONO2/NOyΣRONO2/NOzReferenceUrbanaVancouver,BC12C3-C6mono,0.25-2.5%NAO’brienetal.,19976C2-C6hydroxyaLaPorte,TexasΣRONO20.0-11%0.0-49%Rosenetal.,2004GraniteBay,CAΣRONO24.3-9.5%NAMurphyetal.,2006RuralaHastings,Ontario12C3–C6mono0.50-3.0%NAO’brienetal.,19954C2–C4hydroxy1C4diaPellston,MichiganC3-C5mono0.0-6.0%NAGrossenbacheretal.,2001IsopreneNitratesOstlingetal.,2001aUC-BFRS,CAΣRONO26.0-37%10-50%Dayetal.,2003,2009BigHills,CAΣRONO217-27%NAMurphyetal.,2006AircraftsEasternUSΣRONO212-20%NAPerringetal.,2009MexicoCityΣRONO210-20%NAPerringetal.,2010CanadianBorealForestΣRONO222%NABrowneetal.,2013aNorthPacificΣRONO23.0-20%NABertrametal.,2013bLondonΣRONO27.0-25%NAAruffoetal.,2014155a.测量时间段为白天(8:00-20:00)或SZA<90°b.测量时间为9:55-14:10(UTC)c.数值为该参数在测量期间的变化范围或中值,NA代表无参数在光照条件下的有机硝酸的生成机制与O3的生成是并行的,在光化学污染严重的时段,有机硝酸的浓度也会处于较高水平。我们选取了观测期间O3连续8h平均浓度高于100微克160/立方米的日期作为污染天(分别是7月10、18、23日),其余为清洁天,分析了NOz各组分与“MissingNOz”的日均变化情况(如图3(b)、3(c))。污染天“MissingNOz”浓度在下午时段出现峰值约5ppbv,表明有机硝酸可能在光化学污染严重的时段会出现浓度高值,这为-6-
中国科技论文在线http://www.paper.edu.cnSOA的生成创造了有利条件,从而可能促进O3和PM2.5双高的污染现象的出现。而在清洁天,NOz的日均浓度变化不明显,浓度峰值出现在夜间且低于2ppbv。165图3NOz组成的浓度日均变化及各组分占比:(a)图表示整个观测期间的浓度日均变化;(b)图表示空气污染阶段的浓度日均变化,污染阶段表示O3连续8h平均浓度超过100微克/立方米(共三天);(c)图表示空气清洁阶段浓度日均变化Fig.3ConcentrationofNOzcomponentsandtheirdistributions:Panel(a)representsthemeandiurnalchangesof170NOzconcentrationduringwholecampaign;Panel(b)representsthemeandiurnalchangesofNOzconcentration3duringpollutedperiods,whicharedefinedbyconsecutiveozoneconcentrationhigherthan100μg/m;Panel(c)representsthemeandiurnalchangesofNOzconcentrationduringcleanperiods2.2RACM2中有机硝酸的生成速率当NOx浓度相对较高时,NO+RO2、NO2+OH是HOx循环的主要终止方式,因此RO2175主要通过NO+RO2去除;而在本次观测期间,NOx浓度相对较低,RO2主要通过RO2+HO2生成过氧化物去除,这表明有机硝酸的生成受到过氧化物反应的抑制。因此定义了一个参数β表示RO2与NO反应在RO2的所有反应中的占比,具体的计算如式1所示:日间有机硝酸的生成速率可以通过计算OH自由基与前体物VOCs反应得到,基于观测得到180的各类VOCs我们可以通过式2计算得到有机硝酸在白天的生成速率:-7-
中国科技论文在线http://www.paper.edu.cn[36][42][57]式2所代表的反应步骤是有机硝酸在白天产生的决速反应,其中[RHi]为实测的第i种VOC浓度,和αi分别表示OH自由基和第i种VOCs的反应速率以及有机硝酸的产率(见表4)。185表4观测的各类VOCs及用来计算P(Ox)、P(∑RONO2)、VOC-OH反应活性所使用的α和kOHTab.4MeasuredVOCswiththeirkOHandαforcalculatingP(Ox),P(∑RONO2),VOC-OHreactivityababSpeciesBranchingratioαkOHSpeciesBranchingratioαkOHAlkanes-15-12Methane0.0016.34×102,2-Dimethylbutane0.1522.34×10-13-12Ethane0.0092.58×102,3-Dimethylbutane0.0615.78×10-12dPropane0.0361.10×102,3-Dimethylpentane---12dn-Butane0.0832.54×102,4-Dimethylpentane---12-12Isobutane0.0272.19×102-Methylpentane0.1115.30×10-12-12n-Pentane0.1234.00×102-Methylhexane0.2056.86×10-12dIsopentane0.0753.90×102-Methylheptane---12-12n-Hexane0.2125.45×103-Methylpentane0.1095.40×10-12-12n-Heptane0.2787.02×103-Methylhexane0.1927.15×10-12dn-Octane0.3468.71×103-Methylheptane---12c-12n-Nonane0.3939.99×10Cyclopentane0.0454.97×10-11c-12n-Decane0.4171.12×10Cyclohexane0.166.97×10-11-12n-Undecane0.4311.29×10Methylcyclopentane0.145.60×10-11-12n-Dodecane0.4401.39×10Methylcyclohexane0.179.64×10Alkenes-12-11Ethene0.00058.20×10cis-2-Butene0.0415.64×10-11-11Propylene0.0212.63×10trans-2-Butene0.0416.40×10-11-111-Butene0.0393.14×10cis-2-Pentene0.0646.70×10-11-111-Pentene0.0593.14×10trans-2-Pentene0.0646.70×10c-10Isoprene0.071.01×10Aromatics-12-11Benzene0.0291.22×10m-Xylene0.0742.3×10-12-11Ethylbenzene0.0727.00×10p-Xylene0.0971.43×10-12dn-Propylbenzene0.0935.80×10p-Diethylbenzene---12dIsopropylbenzne0.116.30×10m-Diethylbenzene---12-11Toluene0.0795.96×10Styrene05.80×10-11-11m-Ethyltoluene0.0941.86×101,2,3-Trimethylbenzene0.1053.27×10-11-11o-Ethyltoluene0.1061.19×101,2,4-Trimethylbenzene0.1053.25×10-11-11p-Ethyltoluene0.1371.18×101,3,5-Trimethylbenzene0.1275.76×10-11o-Xylene0.0811.36×10a.VOCs的有机硝酸产率(Branchingratiosα)来自MasterChemicalMechanism:http://mcm.leeds.ac.uk/MCM/b.VOCs的OH自由基反应活性kOH来自MasterChemicalMechanism(T=298K)[58]c.αandkOH的值来自(Lockwood等,2010)-12190d.在MCM中无该VOCs的相关参数,本研究的计算中使用α=0.05和kOH=1.0×10通过比对式1计算得到的有机硝酸生成速率(称为计算值)与基于RACM2机理模拟得到的有机硝酸生成速率(称为模拟值),可以判断RACM2机理对有机硝酸生成机制描述的可靠性。两者的比对结果如图4所示,蓝线代表模拟值P(∑RONO2)Model,而绿点表示计算值P(∑-8-
中国科技论文在线http://www.paper.edu.cnRONO2)Calc。模拟值与计算值的有机硝酸生成速率在变化趋势上基本一致,均在正午到下午195时段出现最大值(峰值范围为0.2-0.7ppbv/h),说明RACM2机理对有机硝酸的生成速率的归并具有较好的代表性,与现行的有机硝酸生成机制基本吻合,但从数值水平来看模拟值比计算值平均高27%左右,主要是由于机理中各类归纳VOC(HC3、HC5、HC8、ISO、BEN、TOL等)的有机硝酸产率α偏高导致。在RACM2机理中,不同VOCs按照其OH自由基反应活性进行分类归纳,而各归纳物200质的有机硝酸产率是根据该归纳物质所包含的实际化学物质的美国源排放强度加权得到的,与本次观测的化学条件不完全一致。为了提高有机硝酸产率的计算精度,我们基于本次观测所得到的VOCs数据,依据RACM2机理中的VOCs分类方式重新加权计算了各归纳物质的有机硝酸产率,修改前和修改后的α值分别用α(RACM2)和α(fixed)表示,如表5所示,原始产率较低的芳香烃类物质经修改后略有提高,而产率较高的长链烷烃如HC8和异戊二烯205ISO则在修改后降低。通过计算光照条件下有机硝酸化学生成来源可知,异戊二烯所氧化的到的有机硝酸在白天占比超过50%,是最主要的贡献源,这与观测地点周围植被覆盖率大,生物源VOC排放显著有关。而HC8与异戊二烯的有机硝酸产率的下降导致P(∑RONO2)Model明显减小。图5表示α值修正前和修正后模拟的有机硝酸生成速率与计算值的相关性分析的结果,虽然修正前后模拟值和计算值的相关性都较强,但α值修正后的模拟值和计算值之间210的斜率为基本为1(≈0.95),说明经过修正后的RAMC2所模拟得到的有机硝酸生成速率与计算值基本吻合,也体现出使用归纳机理对有机硝酸的模拟中α值本地化的重要性。图4观测期间(太阳高度角小于90°)P(∑RONO2)Model与P(∑RONO2)Calc的变化情况,其中蓝线表示模拟得到的P(∑RONO2)Model、绿点表示P(∑RONO2)Calc215Fig.4TimeseriesofP(∑RONO2)Model(bluelines)andP(∑RONO2)Calc(greendots)whenSZA<90°.表5RACM2中各VOC归纳物的有机硝酸原始产率及经过观测浓度修正后的产率Tab.5BranchingratiosofVOCssurrogatesinRACM2beforeandaftertheyarefixedVOC(RACM2)α(RACM2)α(fixed)XYO0.050.085XYM0.050.093XYP0.050.134TOL0.050.05BEN0.0290.029-9-
中国科技论文在线http://www.paper.edu.cnOLI0.060.047OLT0.040.041HC30.0650.059HC50.1360.158HC80.2480.231ISO0.120.07220图5基于VOC观测结果计算和RACM2模拟得到的有机硝酸生成速率相关性(红色点表示RACM2机理中有机硝酸产率为原始值,基于美国排放清单得到;蓝色点表示RACM2机理中的有机硝酸产率经过我国实际观测浓度的修正)Fig.5LinearregressionforcalculatingorganicnitrateproductionbasedonspecificVOCmeasurementsandmodelresultsbasedonRACM2(redpointsrepresenttheresultbeforebranchingratiosarefixed;bluepointsrepresentthe225resultafterbranchingratiosarefixed)2.3αeff与臭氧生成的抑制从反应式R3、R4可知,对于不同的VOC前体物均存在相对应的RONO2反应途径占比α,剩余的1-α通道则会生成NO2,RO也会继续与O2、NO反应生成NO2,并最终光解产生O3。对于一般的VOC而言,1分子的VOC能够导致2分子的NO转化成NO2或生成1分230子的RONO2,少数VOC的后续反应则能导致更多的NO被氧化为NO2。若将混合VOCs气团看作由一类特定的VOC组成,则该气团也有其自身的产率α,定义为有效有机硝酸产率αeff。在之前的研究中,αeff的计算方式为:上式中和分别表示和的浓度变化(用Ox=O3+NO2代替O3能更好235的代表R3的反应速率),表示VOCs能够产生的NO2分子数。该计算方式只有在O3、RONO2[36]的去除、传输作用相对于其生成作用可以忽略时才能成立。而本次观测中由于缺少有机硝酸浓度的观测值,且对于臭氧的传输作用没有定量,因此在本研究中对αeff的计算采用:其中,与均是通过模型模拟得到的速率值,该方法与Browne等(2012)中对240αeff的计算方式类似。观测期间的αeff在太阳天顶角<90°的时间段内中值为0.033,日变化(仅对太阳天顶角<90°的时间段计算)如图6所示,可以发现其平均值从日出前的0.03爬升至日出后的0.04,-10-
中国科技论文在线http://www.paper.edu.cn主要原因可能是在日出后生物源排放大量BVOCs,这些BVOCs的有机硝酸产率较夜间积累的小分子VOCs高,使得αeff升高。纵观整个观测期间,该参数值最大变化幅度接近0.06。245由图6可以看到,同样在乡村地区观测计算得到的αeff值中,MexicoCity和Texa州的αeff[36][11][34]高于本次观测值,而加州(CA)的观测值则略低,总的来说本次观测中的αeff处于中等水平,在部分时段有高值(约0.06)出现,说明了在OH自由基氧化VOCs的过程中,臭氧的生成在部分时段被抑制得较为显著。另外,有研究指出即使是低有机硝酸产率的VOCs对[45]O3生成的抑制作用也不可忽略。250图6观测期间αeff的日变化箱式图(仅对太阳天顶角<90°进行计算),其中红点代表均值。A,MexicoCity(Farmer等,2011);B,LaPorte,Texas的早晨(09:00-12:00)与下午(14:00-18:00)平均(Rosen等,2004);C,UC-BFRS,CA(Day等,2003)Fig.6Boxplotofαeffdiurnalchangeduringthecampaign(onlywhenSZA<90°).Redpointsshowmeandiurnal255change.A,MexicoCity(Farmeretal.,2011);B,LaPorte,meanvaluesofmornings(09:00-12:00)andafternoons(14:00-18:00)atTexas(Rosenetal.,2004);C,UC-BFRS,CA(Dayetal.,2003)由表3可以得到,不同的VOCs分子具有不同的有机硝酸产率α,导致气团的有机硝酸有效产率αeff与其VOCs的组成和浓度水平密切相关,当VOCs中普遍以高分子量的VOCs为主时,αeff较高,当VOCs中普遍以低分子量的VOCs为主时,αeff较低。如图7(a)所示,2260我们发现观测期间气团的VOC-OH反应活性与αeff呈现较好的正相关(R=0.435),主要原因是气团的VOCs组成发生明显变化。当气团中主要以低分子VOCs为主时,整体的VOC-OH反应活性与αeff都偏低,当气团转变为以高分子VOCs为主时,其VOC-OH反应活性与αeff都会升高。在气团组分由小分子VOCs主导逐渐转变为由高分子VOCs主导的过程中,VOC-OH265反应活性与αeff同时升高,使得臭氧的生成受到有机硝酸生成的抑制作用加强,从而出现如图7(b)所示的P(Ox)随VOC-OH反应活性变化的规律(8ppbv<[NOx]<12ppbv),即在VOC-OH反应活性不断升高的过程中,P(Ox)先快速升高至峰值,后缓慢下降至接近零,这与Farmer[11]等人的研究结果类似。利用臭氧生成速率的决速步骤计算公式(式5)可以更好的理解上述现象:270上式中将VOCs气团看作一个整体,代表气团中的VOC分子平均可以产生的NO2分子数,该值一般为2,在下述分析中我们假定其为常数;的计算和定义见3.2;为整个气团的VOC-OH反应活性;为OH自由基的浓度,该计算式与之前的研究中所使[36][57][59]用的计算方式类似。假设在NOx浓度不发生改变的情况下,当气团中VOC组分发275生转变,VOC-OH反应活性不断升高即增大时,与变小,使得P(Ox)-11-
中国科技论文在线http://www.paper.edu.cn[60]会在某个位置达到峰值。这与经典的EKMA曲线所描述的P(Ox)变化规律不同,在传统的EKMA分析中,前提假设是VOCs的组分固定,因而αeff也为定值。2图7(a)气团VOC活性与αeff之间的相关关系(R=0.435);(b)P(Ox)随VOC-OH反应活性变化趋势280(8ppbv<[NOx]<12ppbv)2Fig.7(a)EffectivebranchingratioofairmassvariesasafunctionofVOC-OHreactivity(R=0.435);(b)P(Ox)variesasafunctionofVOC-OHreactivity(8ppbv<[NOx]<12ppbv)[61]在研究臭氧生成机制时,很少有研究者会注意有机硝酸生成对其的抑制作用,本研究表明随着VOCs气团组分的改变,其有机硝酸的光化学生成对臭氧生成的抑制作用也会发285生变化。当臭氧的生成处于P(Ox)随VOC-OH活性的升高而降低的区域时,削减高分子量的VOCs会导致P(Ox)升高。这一结果预示着在削减VOCs控制臭氧时,需要视情况来判定削减高分子量还是低分子量的VOCs,从而有效的降低P(Ox)。3.结论(1)在珠三角的大型综合观测(PRIDE-PRD2006)中发现“missingNOz”占比较高接近29040%,表明珠三角地区可能存在高浓度的有机硝酸。同时在臭氧污染天,“missingNOz”浓度较清洁天高,且出现明显的午间高峰,这与臭氧浓度变化情况类似。也表明日间可能由较高浓度水平的有机硝酸存在。(2)利用得到的VOCs与OH自由基浓度数据,根据有机硝酸决速步骤计算得到观测期间总有机硝酸生成速率P(ΣRONO2)calc,将其与盒子模型耦合RACM2机理模拟得到的2295P(ΣRONO2)model进行比对发现虽然在变化趋势上吻合(R=0.93),但模拟值高于计算值。随后通过将机理中的有机硝酸产率根据本次观测结果进行本地化后,得到的模拟值与计算值大小吻合较好,斜率接近1,说明在使用归纳机理模拟有机硝酸生成时,需要将机理中归纳物质的产率进行本地化。(3)通过模式模拟有机硝酸与臭氧的化学生成速率可计算得到气团的有效有机硝酸产率αeff300(约为0.033),相较于其他研究,本观测期间的αeff值处于中等水平。另外,气团中VOCs组-12-
中国科技论文在线http://www.paper.edu.cn分的改变是使VOC-OH反应活性与αeff存在正相关关系的主要因素,也因此P(Ox)随着VOC-OH反应活性呈现先升高后降低的变化趋势。故在珠三角地区考虑控制VOCs浓度来削减臭氧浓度时,应视不同情况制定相应的VOC控制策略。在高VOCs活性的城市及周边地区,应该优先考虑低分子量VOCs的削减,从而有效抑制臭氧污染。305[参考文献](References)[1]ITOA,SILLMANS,PENNERJE.Globalchemicaltransportmodelstudyofozoneresponsetochangesinchemicalkineticsandbiogenicvolatileorganiccompoundsemissionsduetoincreasingtemperatures:Sensitivities310toisoprenenitratechemistryandgridresolution[J].JGeophysRes,2009,114(D9).[2]BROWNEE,COHENR.EffectsofbiogenicnitratechemistryontheNOxlifetimeinremotecontinentalregions[J].AtmosChemPhys,2012,12(24):11917-32.[3]KHANM,COOKEM,UTEMBES,etal.GlobalmodelingoftheC1-C3alkylnitratesusingSTOCHEM-CRI[J].AtmosEnviron,2015,123:256-267.315[4]CHENX,HULBERTD,SHEPSONPB.MeasurementoftheorganicnitrateyieldfromOHreactionwithisoprene[J].JGeophysRes,1998,103(D19):25,563-25,568.[5]BERTRAMT,PERRINGA,WOOLDRIDGEP,etal.OntheexportofreactivenitrogenfromAsia:NOxpartitioningandeffectsonozone[J].AtmosChemPhys,2013,13(9):4617-4630.[6]LIANGJ,HOROWITZLW,JACOBDJ,etal.Seasonalbudgetsofreactivenitrogenspeciesandozoneover320theUnitedStates,andexportfluxestotheglobalatmosphere[J].JGeophysRes,1998,103(D11):13435-13450.[7]ROMERPS,DUFFEYKC,WOOLDRIDGEPJ,etal.Thelifetimeofnitrogenoxidesinanisoprene-dominatedforest[J].AtmosChemPhys,2016,16(12):7623-7637.[8]CALVERTJ,MADRONICHS.Theoreticalstudyoftheinitialproductsoftheatmosphericoxidationofhydrocarbons[J].JGeophysRes,1987,92(D2):2211-2220.325[9]O"BRIENJM,CZUBAE,HASTIEDR,etal.DeterminationoftheHydroxyNitrateYieldsfromtheReactionofC2−C6AlkeneswithOHinthePresenceofNO[J].JPhysChemA,1998,102(45):8903-8908.[10]FLOCKEF,VOLZ‐THOMASA,BUERSHJ,etal.Long‐termmeasurementsofalkylnitratesinsouthernGermany:1.Generalbehaviorandseasonalanddiurnalvariation[J].JGeophysRes,1998,103(D5):5729-46.[11]FARMERD,PERRINGA,WOOLDRIDGEP,etal.Impactoforganicnitratesonurbanozoneproduction[J].330AtmosChemPhys,2011,11(9):4085-94.[12]ARUFFOE,CARLOPD,DARI-SALISBURGOC,etal.Aircraftobservationsofthelowertroposphereaboveamegacity:Alkylnitrateandozonechemistry[J].AtmosEnviron,2014,94:479-488.[13]ROLLINSAW,KIENDLER-SCHARRA,FRYJ,etal.Isopreneoxidationbynitrateradical:alkylnitrateandsecondaryorganicaerosolyields[J].AtmosChemPhys,2009,9(18):6685-6703.335[14]ROLLINSA,BROWNEE,MINK-E,etal.EvidenceforNOxcontrolovernighttimeSOAformation[J].Science,2012,337(6099):1210-1212.[15]DAYDA,LIUS,RUSSELLLM,etal.OrganonitrategroupconcentrationsinsubmicronparticleswithhighnitrateandorganicfractionsincoastalsouthernCalifornia[J].AtmosEnviron,2010,44(16):1970-1979.[16]KIENDLER‐SCHARRA,MENSAHA,FRIESEE,etal.Ubiquityoforganicnitratesfromnighttime340chemistryintheEuropeansubmicronaerosol[J].GeophysResLett,2016,43(14):7735-7744.[17]AREYJ,ASCHMANNSM,KWOKES,etal.AlkylNitrate,HydroxyalkylNitrate,andHydroxycarbonylFormationfromtheNOx−AirPhotooxidationsofC5−C8n-Alkanes[J].JPhysChemA,2001,105(6):1020-7.[18]SIMPSONIJ,WANGT,GUOH,etal.Long-termatmosphericmeasurementsofC1-C5alkylnitratesinthePearlRiverDeltaregionofsoutheastChina[J].AtmosEnviron,2006,40(9):1619-32.345[19]ARCHIBALDA,KHANM,WATSONL,etal.Commenton"Long-termatmosphericmeasurementsofC1-C5alkylnitratesinthePearlRiverDeltaregionofsoutheastChina"bySimpsonetal[J].AtmosEnviron,2007,41(34):7369-70.[20]ROLLINSAW,SMITHJD,WILSONKR,etal.Realtimeinsitudetectionoforganicnitratesinatmosphericaerosols[J].EnvironSciTechnol,2010,44(14):5540-5.350[21]FRYJ,KIENDLER-SCHARRA,ROLLINSA,etal.OrganicnitrateandsecondaryorganicaerosolyieldfromNO3oxidationofβ-pineneevaluatedusingagas-phasekinetics/aerosolpartitioningmodel[J].AtmosChemPhys,2009,9(4):1431-49.[22]AYRESB,ALLENH,DRAPERD,etal.OrganicnitrateaerosolformationviaNO3+biogenicvolatileorganiccompoundsinthesoutheasternUnitedStates[J].AtmosChemPhys,2015,15(23):13377-92.355[23]FAHEYD,HüBLERG,PARRISHD,etal.Reactivenitrogenspeciesinthetroposphere:MeasurementsofNO,NO2,HNO3,particulatenitrate,peroxyacetylnitrate(PAN),O3,andtotalreactiveoddnitrogen(NOy)atNiwotRidge,Colorado[J].JGeophysRes,1986,91(D9):9781-93.[24]ATLASE.Evidencefor≥C3alkylnitratesinruralandremoteatmospheres[J].Nature,1988,331(6155):426-8.-13-
中国科技论文在线http://www.paper.edu.cn360[25]BOTTENHEIMJW,BARRIELA,ATLASE.ThepartitioningofnitrogenoxidesinthelowerArctictroposphereduringspring1988[J].JAtmosChem,1993,17(1):15-27.[26]O"BrienJM,ShepsonPB,MuthuramuK,etal.MeasurementsofalkylandmultifunctionalorganicnitratesataruralsiteinOntario[J].JournalofGeophysicalResearchAtmospheres,1995,100(D11):22795-22804.[27]O"BRIENJ,SHEPSONP,WUQ,etal.Productionanddistributionoforganicnitrates,andtheirrelationship365tocarbonylcompoundsinanurbanenvironment[J].AtmosEnviron,1997,31(14):2059-69.[28]O"BRIENJM,CZUBAE,HASTIEDR,etal.DeterminationoftheHydroxyNitrateYieldsfromtheReactionofC2−C6AlkeneswithOHinthePresenceofNO[J].JPhysChemA,1998,102(45):8903-8.[29]LUXENHOFERO,SCHNEIDERM,DAMBACHM,etal.SemivolatilelongchainC6-C17alkylnitratesastracecompoundsinair[J].Chemosphere,1996,33(3):393-404.370[30]SCHNEIDERM,LUXENHOFERO,DEISSLERA,etal.C1−C15AlkylNitrates,BenzylNitrate,andBifunctionalNitrates:MeasurementsinCaliforniaandSouthAtlanticAirandGlobalComparisonUsingC2Cl4andCHBr3asMarkerMolecules[J].EnvironSciTechnol,1998,32(20):3055-62.[31]GROSSENBACHERJW,COUCHT,SHEPSONPB,etal.Measurementsofisoprenenitratesaboveaforestcanop[J].JGeophysRes,2001,106(D20):24429-38.375[32]TREVESK,RUDICHY.TheAtmosphericFateofC3−C6HydroxyalkylNitrates[J].JPhysChemA,2003,107(39):7809-17.[33]DAYD,WOOLDRIDGEP,DILLONM,etal.Athermaldissociationlaser‐inducedfluorescenceinstrumentforinsitudetectionofNO2,peroxynitrates,alkylnitrates,andHNO3[J].JGeophysRes,2002,107(D6):4064.380[34]DAYDA,DILLONMB,WOOLDRIDGEPJ,etal.Onalkylnitrates,O3,andthe“missingNOy”[J].JGeophysRes,2003,108(D16)。[35]DAYD,FARMERD,GOLDSTEINA,etal.ObservationsofNOx,ΣPNs,ΣANs,andHNO3ataruralsiteintheCaliforniaSierraNevadaMountains:summertimediurnalcycles[J].AtmosChemPhys,2009,9(14):4879-96.[36]ROSENR,WOODE,WOOLDRIDGEP,etal.ObservationsoftotalalkylnitratesduringTexasAirQuality385Study2000:ImplicationsforO3andalkylnitratephotochemistry[J].JGeophysRes,2004,109(D7).[37]MURPHYJ,DAYD,CLEARYP,etal.ObservationsofthediurnalandseasonaltrendsinnitrogenoxidesinthewesternSierraNevada[J].AtmosChemPhys,2006,6(12):5321-38.[38]PAULD,FURGESONA,OSTHOFFHD.Measurementsoftotalperoxyandalkylnitrateabundancesinlaboratory-generatedgassamplesbythermaldissociationcavityring-downspectroscopy[J].RevSciInstrum,2009,39080(11):114101.[39]SobanskiN,SchuladenJ,SchusterG,etal.A5-channelcavityring-downspectrometerforthedetectionofNO2,NO3,N2O5,totalperoxynitratesandtotalalkylnitrates[J].AtmosMeasTech,2016,9(10):1-32.[40]SobanskiN,ThieserJ,SchuladenJ,etal.Day-andNight-timeFormationofOrganicNitratesataForestedMountain-siteinSouthWestGermany[J].AtmosChemPhys,2017:1-26.395[41]ThieserJ,SchusterG,PhillipsGJ,etal.Atwo-channel,ThermalDissociationCavity-RingdownSpectrometerforthedetectionofambientNO2,RO2NO2andRONO2[J].Genomics,2017,8(11):11533-11596.[42]PERRINGA,BERTRAMT,FARMERD,etal.TheproductionandpersistenceofΣRONO2intheMexicoCityplume[J].AtmosChemPhys,2010,10(15):7215-29.[43]ITOA,SILLMANS,PENNERJE.Effectsofadditionalnonmethanevolatileorganiccompounds,organic400nitrates,anddirectemissionsofoxygenatedorganicspeciesonglobaltroposphericchemistry[J].JGeophysRes,2007,112(D6).[44]XIEY,ELLEMANR,JOBSONT,etal.EvaluationofO3‐NOx‐VOCsensitivitiespredictedwiththeCMAQphotochemicalmodelusingPacificNorthwest2001fieldobservations[J].JGeophysRes,2011,116(D20).[45]LINGZ,GUOH,SIMPSONIJ,etal.Newinsightintothespatiotemporalvariabilityandsource405apportionmentsofC1-C4alkylnitratesinHongKong[J].AtmosChemPhys,2016,16(13):8141-56.[46]LOUS,HOLLANDF,ROHRERF,etal.AtmosphericOHreactivitiesinthePearlRiverDelta-Chinainsummer2006:measurementandmodelresults[J].AtmosChemPhys,2010,10(22):11243-60.[47]LUK,ROHRERF,HOLLANDF,etal.ObservationandmodellingofOHandHO2concentrationsinthePearlRiverDelta2006:amissingOHsourceinaVOCrichatmosphere[J].AtmosChemPhys,2012,12(3):4101541-69.[48]ZHANGJ,WANGT,CHAMEIDESW,etal.OzoneproductionandhydrocarbonreactivityinHongKong,SouthernChina[J].AtmosChemPhys,2007,7(2):557-73.[49]CHANCK,YAOX.AirpollutioninmegacitiesinChina[J].AtmosEnviron,2008,42(1):1-42.[50]PERRINGA,BERTRAMT,WOOLDRIDGEPJ,etal.AirborneobservationsoftotalRONO2:new415constraintsontheyieldandlifetimeofisoprenenitrates[J].AtmosChemPhys,2009,9(4):1451-63.[51]BUHRMP,PARRISHDD,NORTONRB,etal.ContributionoforganicnitratestothetotalreactivenitrogenbudgetataruraleasternUSsite[J].JGeophysRes,1990,95(D7):9809-16.[52]PARRISHD,BUHRM,TRAINERM,etal.ThetotalreactiveoxidizednitrogenlevelsandthepartitioningbetweentheindividualspeciesatsixruralsitesineasternNorthAmerica[J].JGeophysRes,1993,98(D2):4202927-39.[53]SANDHOLMS,OLSONJ,BRADSHAWJ,etal.SummertimepartitioningandbudgetofNOycompoundsinthetroposphereoverAlaskaandCanada:ABLE3B[J].JGeophysRes,1994,99(D1):1837-61.[54]HAYDENK,ANLAUFK,HASTIED,etal.PartitioningofreactiveatmosphericnitrogenoxidesatanelevatedsiteinsouthernQuebec,Canada[J].JGeophysRes,2003,108(D19).425[55]HORIICV,MUNGERJW,WOFSYSC,etal.AtmosphericreactivenitrogenconcentrationandfluxbudgetsataNortheasternUSforestsite[J].AgricForMeteorol,2005,133(1):210-25.-14-
中国科技论文在线http://www.paper.edu.cn[56]OSTLINGK,KELLYB,BIRDS,etal.Fast‐turnaroundalkylnitratemeasurementsduringthePROPHET1998summerintensive[J].JGeophysRes,2001,106(D20):24439-49.[57]BROWNEE,MINK-E,WOOLDRIDGEP,etal.ObservationsoftotalRONO2overtheborealforest:NOx430sinksandHNO3sources[J].AtmosChemPhys,2013,13(9):4543-62.[58]LOCKWOODA,SHEPSONP,FIDDLERM,etal.Isoprenenitrates:preparation,separation,identification,yields,andatmosphericchemistry[J].AtmosChemPhys,2010,10(13):6169-78.[59]PERRINGA,PUSEDES,COHENR.Anobservationalperspectiveontheatmosphericimpactsofalkylandmultifunctionalnitratesonozoneandsecondaryorganicaerosol[J].ChemRev,2013,113(8):5848-70.435[60]唐孝炎,张远航,邵敏.大气环境化学(第二版)[M].北京:高等教育出版社,2006.[61]WANGT,XUEL,BRIMBLECOMBEP,etal.OzonepollutioninChina:Areviewofconcentrations,meteorologicalinfluences,chemicalprecursors,andeffects[J].SciTotalEnviron,2017,575:1582-1596.-15-'
您可能关注的文档
- 山前带复杂构造偏移成像影响因素分析.pdf
- 柳珊瑚内生真菌Aspergillus versicolor的天然产物研究.pdf
- 柴达木盆地昆北地区路乐河组下干柴沟组泥岩地层地球化学及古环境意义.pdf
- 氮杂环丁烷衍生的锌配合物合成及催化研究.pdf
- 水稻XYLP7基因在维管系统发育中的功能.pdf
- 浪致混合效应的耦合模式模拟.pdf
- 海绵状石墨烯镍颗粒混合结构可拉伸气敏传感器的制备.pdf
- 潜在蒸散发数据对网格HBV模型降雨径流模拟的影响分析.pdf
- 烯丙基荧光素-丙烯酰胺沉淀共聚荧光分子印迹选择性荧光检测三氟氯氰菊酯残留.pdf
- 石墨烯分散液光限幅特性的温度调控.pdf
- 竹笋壳基活性炭材料的制备及其超级电容性能研究.pdf
- 莱维飞行中的轨线动力学.pdf
- 阿勒泰泥炭重金属元素异常记录的人类活动信息.pdf
- 陕西双王金矿床中自然金属与金属互化物.pdf
- 预磁化对SmBCO超导块材磁悬浮性能的影响.pdf
- 高效液相色谱法测定野生和人工种植库鲁木提草中柚皮苷和木犀草素的含量.pdf
- 高能球磨法对Li3Mg2NbO6陶瓷结构及性能的影响.pdf
- 1,25(OH)2D3对谷氨酸诱导HT-22细胞损伤保护作用研究.pdf
相关文档
- 施工规范CECS140-2002给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程
- 施工规范CECS141-2002给水排水工程埋地钢管管道结构设计规程
- 施工规范CECS142-2002给水排水工程埋地铸铁管管道结构设计规程
- 施工规范CECS143-2002给水排水工程埋地预制混凝土圆形管管道结构设计规程
- 施工规范CECS145-2002给水排水工程埋地矩形管管道结构设计规程
- 施工规范CECS190-2005给水排水工程埋地玻璃纤维增强塑料夹砂管管道结构设计规程
- cecs 140:2002 给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程(含条文说明)
- cecs 141:2002 给水排水工程埋地钢管管道结构设计规程 条文说明
- cecs 140:2002 给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程 条文说明
- cecs 142:2002 给水排水工程埋地铸铁管管道结构设计规程 条文说明