

- 220.06 KB
- 2022-04-22 13:36:40 发布


- 1、本文档共5页,可阅读全部内容。
- 2、本文档内容版权归属内容提供方,所产生的收益全部归内容提供方所有。如果您对本文有版权争议,可选择认领,认领后既往收益都归您。
- 3、本文档由用户上传,本站不保证质量和数量令人满意,可能有诸多瑕疵,付费之前,请仔细先通过免费阅读内容等途径辨别内容交易风险。如存在严重挂羊头卖狗肉之情形,可联系本站下载客服投诉处理。
- 文档侵权举报电话:19940600175。
'HG3627-1999前言抓氛菊醋是人工合成的拟除虫菊醋类杀虫剂,具有触杀和胃毒作用,杀虫谱广,广泛应用于农业和卫生等领域。本标准技术要求参照联合国粮农组织(FAO)农药规格[FAOSpecification332/TC/S/F(1993)],对抓佩菊醋原药的外观、抓氛菊醋总含量、水分、酸度项目作了规定,指标与之接近;同时将FAO农药规格中顺式异构体含量指标改为高效、低效异构体比,增加了保证期。本标准中抓氛菊醋总含量的测定方法参照采用国际农药分析合作理事会CIPAC332/TC/(M)/3.2,将其中的流动相由IF(异辛烷:乙酸乙醋)=99.5e0.5改为lY(正己烷:乙酸乙酷)=99:1,本标准自实施之日起,代替化工行业标准HG/T2987-1988((抓氛菊醋含量分析方法》。本标准的附录A是标准的附录。本标准由国家石油和化学工业局政策法规司提出。本标准由沈阳化工研究院技术归口。本标准主要起草单位:沈阳化工研究院。本标准参加起草单位:天津龙灯化工有限公司、南京第一农药厂、江苏扬农化工集团有限公司。本标准主要起草人:许来威、张雪冰、楼少挽、邹立冬、樊文中、王锉忠。1238
中华人民共和国化工行业标准HG3627-1999撅橄菊酉旨原药代替HG/T2987-1988Cypermethrintechnical抓氛菊醋的其他名称、结构式和基本物化参数如下:ISO通用名称:CypermethrinCIPAC数字代号:332化学名称:(RS)-a-氛基一3-苯氧节基(IRS)-顺式一反式一3-(2,2一二抓乙烯基-2,2一二甲基环丙烷梭酸醋结构式:OChC}H-CH---A-11-411}--4J/-CH}}冶CCN/CH,CH.,实验式;CzzH,,C1zNO3相对分子质f:416.31(按1993年国际相对原子质a计)生物活性:杀虫燕气压(200C):190nPa溶解度(g/L,200C):水中为IX10-"^-2X10-";己烷103;乙醇337,丙酮、三抓甲烷、环己酮、二甲苯大于450稳定性:在220"C、中性或酸性条件下稳定,pH=4时最佳,在土城中降解1范围本标准规定了抓佩菊酸原药的要求、试验方法以及标志、标签、包装和贮运。本标准适用于由抓佩菊醋及其生产中产生的杂质组成的抓氛菊醋原药。2引用标准下列标准所包含的条文,通过在本标准中引用而构成为本标准的条文。本标准出版时,所示版本均为有效。所有标准都会被修订,使用本标准的各方应探讨使用下列标准最新版本的可能性。GB/T601-1988化学试剂摘定分析(容A分析)用标准溶液的制备GB/T1600-1979(1989)农药水分测定方法GB/T1604-1995商品农药验收规则GB/T1605-19790989)商品农药采样方法GB3796-1983农药包装通则GB4838-1984乳油农药包装GB/T9008-1988液相色谱法术语柱液相色谱法和平面伍谱M-国家石油和化学工业局1999-06-16批准2000一06一01rHfi1239
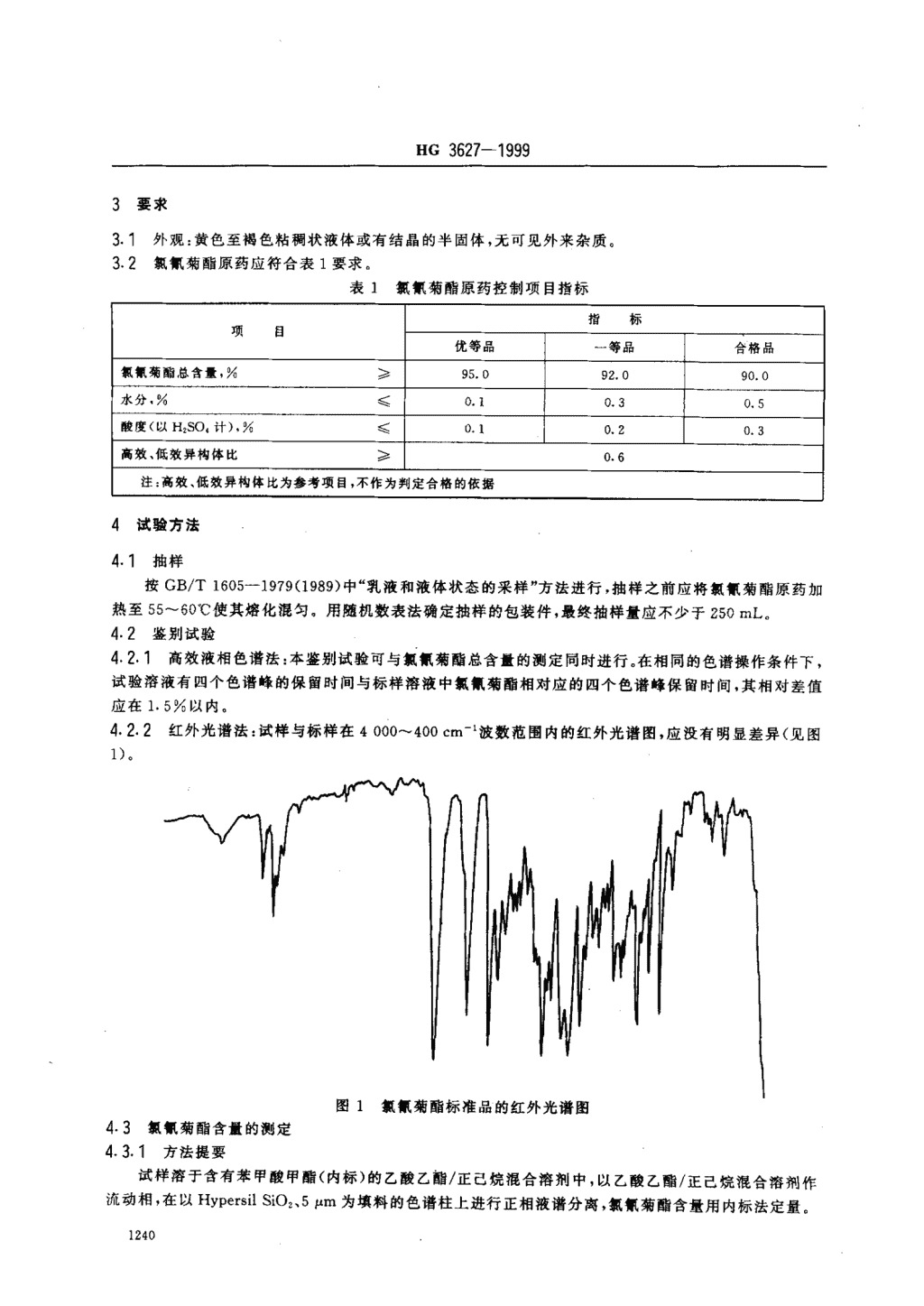
HG3627一19993要求外观:黄色至褐色粘稠状液体或有结晶的半固体,无可见外来杂质。;,;抓佩菊醋原药应符合表1要求。表1抓氛菊酷原药控制项目指标指标项目优等品一等品合格品氛氛菊酣总含量,%》95.092090‘0水分,%(0lO305酸度(以H多0。计),%镇0.10.20。3I高效、低效异构体比李0.6注:高效、低效异构体比为参考项目,不作为判定合格的依据4试验方法4.1抽样按GB/T1605一1979(1989)中“乳液和液体状态的采样”方法进行,抽样之前应将抓氛菊醋原药加热至5一60℃使其熔化混匀。用随机数表法确定抽样的包装件,最终抽样量应不少于250mL。4.2鉴别试验42.1高效液相色谱法:本鉴别试验可与抓佩菊醋总含t的测定同时进行。在相同的色谱操作条件下,试验溶液有四个色谱峰的保留时间与标样溶液中抓佩菊醋相对应的四个色谱峰保留时间,其相对差值应在15鲜以内。闷.22红外光谱法:试样与标样在400~40cm一’波数范围内的红外光谱图,应没有明显差异(见图1)图1抓氛菊醋标准品的红外光谱图4.3抓倾菊醋含最的侧定4.31方法提要试样溶于含有苯甲酸甲醋(内标)的乙酸乙酿/正己烷混合溶剂中,以乙酸乙酷/正己烷混合溶剂作流动相,在以HyPersilsiO:、5拌m为填料的色谱柱上进行正相液谱分离,抓氛菊醋含量用内标法定量。1240
HG3627-19994.3.2仪器、设备液相色谱仪:具有紫外可变波长检测器。色谱数据处理机色谱柱:4.6mm(id)X200mm不锈钢柱,内装HypersilSiO25pm填充物。过滤器:滤膜孔径约。45pm.微量进样器:50pL.4.3.3试剂和溶液正己烷:色谱级。乙酸乙醋:色谱级。流动相:lp(正己烷:乙酸乙醋)=99:1。用移液管移取10mL乙酸乙醋置于990mL正己烷中,摇匀,经0.45pm过滤膜过滤,超声15min,苯甲酸甲酣:应不含有干扰分析的杂质。内标溶液:称取3.8g苯甲酸甲醋,于1000mL容量瓶中,用流动相溶解并定容,摇匀。抓氛菊醋标准品:已知含量,大于等于98.0%.4.3.4液相色谱操作条件柱温:室温(温差变化应不大于2℃)。流动相流量;1.0mL/min,检测波长:278nm,进样体积:10pL,保留时间:苯甲酸甲醋4.5min;低效顺式[(R)-a,(1R)一顺式+(S)-a,(1S)一顺式〕7.8min;高效顺式[(S)-a,(1R)一顺式+(R)-a,(1S)一顺式〕9.2min;低效反式[(R)-a,OR)-反式+(S)-a,0S)-反式〕10.3min;高效反式[(S)-a,(1R)-反式+(R)-a,(1S)-反式〕11.9min(见图2)e1一内标物(苯甲酸甲西)移a-低效顺式[(R)-a,(1R)-顺式+(5卜a,(15)一顺式];b一高效顺式[(S)-a,(1R)-顺式+(R)-a,(1S)-顺式〕,c-低效反式[(R)-a,(1R)-反式+(S)-a,(1S)-反式7;d-高效反式[(S)-a,(1R)-反式+(R)-a,(1S)-反式口图2抓氛菊酸原药液相色谱图上述液相色谱操作条件,系典型操作参数。可根据不同仪器特点,对给定的操作参数作适当调整,以期获得最佳效果。4.3.5M定步骤a)标样溶液的制备称取抓佩菊醋标样0.05g梢确至0.0002g),于15mL具塞玻璃瓶中,用移液管加人10mL内标溶液,溶解摇匀。1241
HG3627-1999b)试样溶液的制备称取含抓氛菊酷0.05g的试样(精确至。.0002g),于15mL具塞玻璃瓶中,用与a)中同一支移液管加人10mL内标溶液,溶解摇匀。再用。.45pm的滤膜过滤。c)测定在上述操作条件下,待仪器稳定后连续注人数针标样溶液,直至相邻两针抓氛菊醋总峰面积与苯甲酸甲醋的峰面积之比的相对变化小于1.0%后,按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进行分析测定。4.3.6计算以质量百分数表示的抓氛菊醋总含量(X,)按式(1)计算:X,一r-Pr1m2式中:不1—标样溶液中抓佩菊醋总峰面积与内标物峰面积之比的平均值;不2—试样溶液中抓氛菊醋总峰面积与内标物峰面积之比的平均值;m,标样的质量,9于,2—试样的质量,9;P—标样中氛氛菊醋的总含量,%。4.3.7允许差取算术平均值作为测定结果。两次平行测定结果之差,应不大于1.5%04.4水分的测定4.4.1测定方法按GB/T160。中的卡尔·费休法进行。允许使用精度相当的水分测定仪测定。4.4.2允许差取算术平均值作为测定结果。两次平行侧定结果之差,应不大于30%.4.5酸度的测定4.5.1试剂和溶液氢氧化钠标准滴定溶液:c(NaOH)二0.02mol/L,按GB/T601规定方法配制。混合指示液:1g/L甲基红乙醇溶液2mL和1g/L浪甲酚绿乙醉溶液10mL混合均匀。95%乙醇。4.5.2测定步骤称取试样2g(精确至。.。。。2g),置于250mL锥形瓶中,加人95%乙醇50mL,振摇使试样溶解均匀、滴加4^-6滴混合指示液,用氢氧化钠标准滴定溶液滴定至由淡红色变为暗绿色为终点。同时作空白测定。4.5.3计算以质量百分数表示的试样的酸度(Xi)(以HZSO。计)按式(2)计算:,,c(V,一V=)X0.049X,一=山=m一X100························⋯⋯(2)式中::—氢氧化钠标准滴定溶液的实际浓度,mot/L;V,滴定试样溶液消耗氮氧化钠标准滴定溶液的体积,mL;VD滴定空白溶液消耗氢氧化钠标准滴定溶液的体积,mL;,—试样的质量,9;0.049—与1.00mL氢氧化钠标准滴定溶液[c(NaOH)=1.000mol/L」相当的以克表示的硫酸的质量。4.5.4允许差1242
HG3627-1999取算术平均值作为测定结果。两次平行测定结果之相对差,应不大于30%.4.6高效、低效异构体比的测定4.6.1测定步骤高效与低效异构体比R的测定可与4.3同时进行。高效与低效异构体比R可按式(3)计算:A6+Ad(3)Aa+A式中:A,-谱峰a的峰面积;A,-谱峰b的峰面积;入—谱峰c的峰面积;Ad-谱峰d的峰面积。4.6.2允许差取算术平均值作为测定结果。两次平行测定结果之差,应不大于0.05,47产品的检验与脸收产品的检验与验收应符合GB/T1604的规定。极限数值的处理采用修约值比较法。5标志、标签、包装、贮运5.1抓氛菊醋原药的标志、标签、包装,应符合GB3796和GB4838中的有关规定。作为商品流通的抓佩菊醋原药,应有生产许可证号和商标。5.2抓氛菊酿原药应用清沽、干操的内衬聚抓乙烯(PVC)的铁桶包装,每桶净含量200kg,5.3根据用户要求或订货协议,可以采用其他形式的包装,但需符合GB4838中的有关规定。5.4抓佩菊酷原药包装件应贮存在通风、千燥的库房中。55贮运时,严防潮湿和日晒,不得与食物、种子、饲料混放,避免与皮肤、眼睛接触,防止由口鼻吸人。5.6安全:本品为菊酌类中等毒性的杀虫剂,可通过皮肤渗人,使用本品应带防护手套、口罩,穿干净防护服。使用后,应立即用肥皂和水洗净。如发现中毒现象,应及时去医院检查治疗。5.7保证期:在规定的贮运条件下,抓氛菊醋原药的保证期,从生产日期算起为2年。1243
HG3627-1999附录A(标准的附录)氮抓菊醋含t的测定A1方法提要试样用正己烷溶解,以正己烷一无水乙醚为流动相,使用以sio:为填料的不锈钢柱和紫外检测器(230nm),对试样中的抓氛菊醋进行正相高效液相色谱分离和测定A2试剂和溶液正己烷:色谱级。无水乙醚:色谱级。流动相:Yp(正己烷:无水乙醚)=98:2,经。.45jam滤膜过滤,并在超声波浴槽中超声10min抓氛菊醋标样:已知含量,大于等于98.0%eA3仪器、设备高效液相色谱仪:具有可变波长紫外检测器。色谱柱:150mmX3.9mm(id)不锈钢柱,内装Nova-PakSiO,填充物,粒径5PM.色谱数据处理机。过滤器:滤膜孔径约为0.5JAM.微量进样器:大于等于50IAL.A4商效液相色谱操作条件流量:1.0mL/min,柱温:室温(温差变化不大于2℃)。检测波长:230nm.进样体积:10I.Lo保留时间:低效顺式[(R)-a,(1R)一顺式+(S)-a,(1S)一顺式〕8.4min;高效顺式[(S)-a,(1R)一顺式+(R)-a,(1S)一顺式〕9.6min;低效反式巨(R)-a,(1R)一反式+(S)-a,(IS)-反式〕10.8min;高效反式〔(S)-a,(1R)-反式+(R)-a,(1S)-反式〕12.3min(见图AD.1244
HG3627-1999abCJUa一低效顺式[(R)-a,(1R)-顺式+(S)-a,(1S)-顺式玉b一高效顺式[(S)-a,(1R)-顺式+(R)-a,(1S)-顺式〕;c一低效反式仁(R)-a,(1R)-反式+(S)-a,(1S)-反式〕;d-高效反式[(S)-a,(1R)-反式+(R)-a,(IS)-反式〕图Al抓氛菊酷原药液相色谱图上述操作参数是典型的,可根据不同仪器特点,对给定操作参数作适当调整,以获得最佳效果(见图Al,AS测定步骤a)标样溶液的配制称取抓氛菊醋标样。.025g(准确至。0002g),置于25mL容量瓶中,用正己烷溶解并稀释至刻度,摇匀。b)试样溶液的配制称取含抓氛菊醋0.025g(准确至。.0002g)的试样,置于25mL容f瓶中,用正己烷溶解并稀释至亥」度,摇匀。c)测定在上述操作条件下,待仪器基线稳定后,连续注人数针标样溶液,直至相邻两针的抓佩菊醋峰面积变化小于1.5%时,按照标样洛液、试样溶液、试样溶液、标样溶液的顺序进行测定。A6计算将测得的两针试样溶液中抓氛菊醋的峰面积以及试样前后两针标样溶液中级氛菊醋的峰面积分别进行平均。以质童百分数表示的抓氛菊醋含a(X,)按式(Al)计算X,““.....··..⋯⋯‘“··········⋯⋯(A1)式中:A,—标样溶液中抓佩菊醋峰面积的平均值A,—试样溶液中饭佩菊酣峰面积的平均值m,-抓氛菊醋标样的质2,9;m2—试样的质量,9;尸—标样中抓氛菊酸的质t百分数,%。A7允许整取其算术平均值作为测定结果。两次平行测定结果之差,应不大于1.0%,1245'
您可能关注的文档
- HG3618-1999苏云金杆菌悬浮剂.pdf
- HG3619-1999仲丁威原药.pdf
- HG3620-1999仲丁威乳油.pdf
- HG3621-1999克百威原药.pdf
- HG3622-19993%克百威颗粒剂.pdf
- HG3623-1999三氯杀虫酯原药.pdf
- HG3624-19992,4-滴原药.pdf
- HG3625-1999丙溴磷原药.pdf
- HG3626-199940%丙溴磷乳油.pdf
- HG3628-1999氯氰菊酯乳油.pdf
- HG3629-1999高效氯氰菊酯原药.pdf
- HG3630-1999高效氯氰菊酯原药浓剂.pdf
- HG3631-19994.5%高效氯氰菊酯乳油.pdf
- HG3634-1999饲料级预糊化淀粉.pdf
- HG3669-2000食品添加剂稳定态二氧化氯溶液.pdf
- HG3671-2000吡虫啉可湿性粉剂.pdf
- HG3672-2000吡虫啉乳油.pdf
- HG3694-2001饲料添加剂乙氧基喹(乙氧基喹啉).pdf
相关文档
- 施工规范CECS140-2002给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程
- 施工规范CECS141-2002给水排水工程埋地钢管管道结构设计规程
- 施工规范CECS142-2002给水排水工程埋地铸铁管管道结构设计规程
- 施工规范CECS143-2002给水排水工程埋地预制混凝土圆形管管道结构设计规程
- 施工规范CECS145-2002给水排水工程埋地矩形管管道结构设计规程
- 施工规范CECS190-2005给水排水工程埋地玻璃纤维增强塑料夹砂管管道结构设计规程
- cecs 140:2002 给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程(含条文说明)
- cecs 141:2002 给水排水工程埋地钢管管道结构设计规程 条文说明
- cecs 140:2002 给水排水工程埋地管芯缠丝预应力混凝土管和预应力钢筒混凝土管管道结构设计规程 条文说明
- cecs 142:2002 给水排水工程埋地铸铁管管道结构设计规程 条文说明